| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 10 mM * 1 mL in DMSO |
|
||
| 1mg |
|
||
| 5mg |
|
||
| 10mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| 500mg |
|
||
| 1g |
|
||
| 2g |
|
||
| Other Sizes |
|
| 靶点 |
JAK2 (IC50 = 5.7 nM); JAK1 (IC50 = 5.9 nM); Tyk2 (IC50 = 53nM); JAK3 (IC50 = 560nM)
|
||
|---|---|---|---|
| 体外研究 (In Vitro) |
基于细胞的研究证明了 baricitinib (INCB028050) 作为 JAK 信号传导和功能抑制剂的效力。 Baricitinib 的 IC50 值分别为 44 nM 和 40 nM,可防止 IL-6 刺激的经典底物 STAT3 (pSTAT3) 磷酸化以及随后在 PBMC 中产生趋化因子 MCP-1。 INCB028050 还在分离的幼稚 T 细胞中抑制 IL-23 激活的 pSTAT3 (IC50=20 nM)。这种抑制可阻止 Th17 细胞产生两种有害细胞因子:IL-17 和 IL-22。 Th17 细胞的 IC50 值为 50 nM,是辅助 T 细胞的亚型,具有独特的炎症和致病特征。即使剂量高达 10 μM,结构相似但无效的 JAK1/2 抑制剂 INCB027753 和 INCB029843 在任何这些测定系统中都没有表现出明显的效果 [1]。
|
||
| 体内研究 (In Vivo) |
在为期 2 周的治疗过程中,baricitinib (INCB028050) 治疗使后爪体积的增加在 1 mg/kg 时减少了 50%,在 3 或 10 mg/kg 时减少了 95% 以上。鉴于爪子体积的基线测量是在治疗第 0 天表现出明显疾病迹象的动物中获得的,那些肿胀明显改善的动物可能表现出 >100% 的抑制作用 [1]。给予巴瑞克替尼(0.7 mg/kg/天)的小鼠表现出 I 类和 II 类 MHC 表达显着降低,CD8 浸润减少,炎症显着减少(通过 H&E 染色测量)。与载体对照动物相比,用巴瑞克替尼治疗的小鼠中 CD8+NKG2D+ 细胞的数量明显较低,CD8+NKG2D+ 细胞是人和小鼠斑秃 (AA) 疾病的重要效应细胞。
|
||
| 酶活实验 |
生化测定[1]
使用具有重组表位标记的激酶结构域(JAK1837-1142;JAK2828-1132;JAK3718-1124;Tyk2873-1187)或全长酶(cMET和Chk2)和肽底物的均匀时间分辨荧光测定法进行酶测定。在测定缓冲液中使用或不使用测试化合物(11点稀释)、JAK、cMET或Chk2酶、500nM(对于Chk2为100nM)肽、ATP(在每种激酶特异性的Km或1mM)和2.0%DMSO进行每种酶反应。计算出的IC50值是抑制50%荧光信号所需的化合物浓度。使用200 nM的标准条件在Cerep进行额外的激酶测定。测试的酶包括:Abl、Akt1、AurA、AurB、CDC2、CDK2、CDK4、CHK2、c-kit、EGFR、EphB4、ERK1、ERK2、FLT-1、HER2、IGF1R、IKKα、IKKβ、JNK1、Lck、MEK1、p38α、p70S6K、PKA、PKCα、Src和ZAP70。 |
||
| 细胞实验 |
细胞测定[1]
通过leukapetheresis和Ficoll-Hypaque离心分离人PBMC。为了测定IL-6诱导的MCP-1的产生,在存在或不存在各种浓度的INCB028050的情况下,将PBMC以3.3×105个细胞/孔的速度接种在RPMI 1640+10%FCS中。在室温下与化合物预孵育10分钟后,通过向每个孔中加入10ng/ml人重组IL-6来刺激细胞。将细胞在37°C、5%CO2下孵育48小时。采集上清液并通过ELISA分析人MCP-1的水平。INCB028050抑制IL-6诱导的MCP-1分泌的能力被报道为50%抑制所需的浓度(IC50)。使用Cell Titer Glo在标准测定条件下在3天内进行Ba/F3-TEL-JAK3细胞的增殖。 为了测定IL-23诱导的IL-17和IL-22,将PBMC维持在补充有10%FBS、2mM l-谷氨酰胺、100μg/ml链霉素和100U/ml青霉素的RPMI 1640培养基中。通过用抗CD3和抗CD28 Abs培养来活化T细胞。2天后,洗涤细胞并用IL-23(100ng/ml)、IL-2(10ng/ml)和各种浓度的INCB028050再培养。将细胞在37°C下再孵育4天,然后收集上清液,并通过ELISA测量IL-17和IL-22的分泌。INCB028050抑制IL-23诱导的IL-17和IL-22分泌的能力被报道为50%抑制所需的浓度(IC50)。 磷酸-STAT3分析[1] 分离的细胞。[1] 为了分析人PBMC或PHA刺激的T细胞中的磷酸化-STAT3,在用不同浓度的INCB028050预孵育10−15分钟并用IL-6(100 ng/ml)、IL-12(20 ng/ml)或IL-23(100 ng/ml)刺激细胞15分钟后制备细胞提取物。然后通过使用磷酸化-STAT3特异性ELISA分析提取物的磷酸化STAT3。 全血。[1] 将从大鼠抽取的血液收集到肝素化管中,然后等分到微量离心管中(每个样品0.3ml)。在刺激实验中,在37°C下用人IL-6(100 ng/ml)刺激15分钟之前,加入不同浓度的INCB028050 10分钟。使用低渗条件裂解RBCs。然后将WBC快速成丸并裂解以制备总的细胞提取物。使用磷酸化STAT3特异性ELISA分析提取物中的磷酸化STAT3。在INCB028050给药后的不同时间从给药INCB02805的动物中抽取血液,并如上所述进行处理。 |
||
| 动物实验 |
|
||
| 药代性质 (ADME/PK) |
Absorption, Distribution and Excretion
The absolute bioavailability of baricitinib is approximately 80%. The Cmax was reached after one hour of oral drug administration. A high-fat meal decreased the mean AUC and Cmax of baricitinib by approximately 11% and 18%, respectively, and delayed Tmax by 0.5 hours. Baricitinib is predominantly excreted via renal elimination. It is cleared via filtration and active secretion. Approximately 75% of the administered dose was eliminated in the urine, with 20% of that dose being the unchanged drug. About 20% of the dose was eliminated in the feces, with 15% of that dose being an unchanged drug. Following intravenous administration, the volume of distribution was 76 L, indicating distribution into tissues. The total body clearance of baricitinib was 8.9 L/h in patients with rheumatoid arthritis. The total body clearance and half-life of baricitinib was 14.2 L/h in intubated patients with COVID-19 who received baricitinib via nasogastric (NG) or orogastric (OG) tube. Metabolism / Metabolites Baricitinib is metabolized by CYP3A4. Approximately 6% of the orally administered dose was identified as metabolites in urine and feces; however, no metabolites of baricitinib were quantifiable in plasma. Biological Half-Life The elimination half-life in patients with rheumatoid arthritis is approximately 12 hours. The elimination half-life was 10.8 hours in intubated patients with COVID-19 who received baricitinib via nasogastric (NG) or orogastric (OG) tube. |
||
| 毒性/毒理 (Toxicokinetics/TK) |
Hepatotoxicity
In the large prelicensure clinical trials in rheumatoid arthritis, serum aminotransferase elevations occurred in up to 17% of baricitinib treated subjects compared to 11% in placebo recipients. The elevations were typically mild and transient and values above 3 times the upper limit of normal (ULN) occurred in 1% to 2% of patients. The elevations occasionally led to early discontinuations, but more often resolved even without dose adjustment. In prelicensure studies in rheumatoid arthritis, alopecia areata and other rheumatic and immune-mediated disorders, there were no instances of clinically apparent liver injury attributed to baricitinib. Since approval and more wide scale availability of baricitinib, there have been no published reports of hepatotoxicity associated with its use. Use of baricitinib in combination with remdesivir for severe COVID-19 pneumonia has been reported but with little information on its potential for causing liver injury. Patients with severe SARS-CoV-2 infection frequently have elevated serum aminotransferase levels and occasionally are jaundiced. Furthermore, remdesivir has been linked to serum aminotransferase elevations during therapy that are generally mild-to-moderate in severity and resolve rapidly once the drug is stopped. Whether baricitinib increases the risk of liver injury during COVID-19 has yet to be shown, but hepatotoxicity was not a prominent feature in these early studies of its use in patients with severe COVID-19. Finally, baricitinib is an immune modulatory agent and has the potential of causing reactivation of viral infections including hepatitis B. In clinical trials, patients with HBsAg in serum were excluded from enrollment but patients with anti-HBc without HBsAg were allowed. While routine monitoring for reactivation was not performed on all patients, at least 15% of anti-HBc positive persons with rheumatoid arthritis treated with baricitinib developed virologic evidence of reactivation marked by de novo appearance of low levels of HBV DNA in serum. In all cases, the period of viremia was brief and not associated with serum aminotransferase elevations or jaundice. Thus, baricitinib appears to be capable of causing HBV reactivation but it is generally subclinical. Furthermore, the short courses of baricitinib used in the treatment of severe COVID-19 have not been linked to episodes of HBV reactivation. Likelihood score: E* (unlikely to be a cause of idiosyncratic clinically apparent liver injury but has the potential to cause reactivation of hepatitis B). Effects During Pregnancy and Lactation ◉ Summary of Use during Lactation No information is available on the use of baricitinib during breastfeeding. Most sources recommend that mothers not breastfeed while taking baricitinib. An alternate drug is preferred, especially while nursing a newborn or preterm infant. The manufacturer recommends that women avoid nursing during therapy and for 4 days after the last dose. ◉ Effects in Breastfed Infants Relevant published information was not found as of the revision date. ◉ Effects on Lactation and Breastmilk Relevant published information was not found as of the revision date. Protein Binding Baricitinib is approximately 50% bound to plasma proteins and 45% bound to serum proteins. |
||
| 参考文献 |
|
||
| 其他信息 |
Pharmacodynamics
Baricitinib is a disease-modifying antirheumatic drug (DMARD) used to ameliorate symptoms and slow down the progression of rheumatoid arthritis. In animal models of inflammatory arthritis, baricitinib was shown to have significant anti-inflammatory effects but also led to the preservation of cartilage and bone, with no detectable suppression of humoral immunity or adverse hematologic effects. Baricitinib decreased the levels of immunoglobulins and serum C-reactive protein in patients with rheumatoid arthritis. |
| 分子式 |
C16H17N7O2S
|
|
|---|---|---|
| 分子量 |
371.42
|
|
| 精确质量 |
371.116
|
|
| 元素分析 |
C, 51.74; H, 4.61; N, 26.40; O, 8.62; S, 8.63
|
|
| CAS号 |
1187594-09-7
|
|
| 相关CAS号 |
Baricitinib phosphate;1187595-84-1;Baricitinib-d5;1564241-79-7;Baricitinib-d3;1564242-30-3
|
|
| PubChem CID |
44205240
|
|
| 外观&性状 |
Typically exists as white to gray solids at room temperature
|
|
| 密度 |
1.6±0.1 g/cm3
|
|
| 沸点 |
707.2±70.0 °C at 760 mmHg
|
|
| 闪点 |
381.5±35.7 °C
|
|
| 蒸汽压 |
0.0±2.3 mmHg at 25°C
|
|
| 折射率 |
1.763
|
|
| LogP |
-0.06
|
|
| tPSA |
128.94
|
|
| 氢键供体(HBD)数目 |
1
|
|
| 氢键受体(HBA)数目 |
7
|
|
| 可旋转键数目(RBC) |
5
|
|
| 重原子数目 |
26
|
|
| 分子复杂度/Complexity |
678
|
|
| 定义原子立体中心数目 |
0
|
|
| SMILES |
S(C([H])([H])C([H])([H])[H])(N1C([H])([H])C(C([H])([H])C#N)(C1([H])[H])N1C([H])=C(C2=C3C([H])=C([H])N([H])C3=NC([H])=N2)C([H])=N1)(=O)=O
|
|
| InChi Key |
XUZMWHLSFXCVMG-UHFFFAOYSA-N
|
|
| InChi Code |
InChI=1S/C16H17N7O2S/c1-2-26(24,25)22-9-16(10-22,4-5-17)23-8-12(7-21-23)14-13-3-6-18-15(13)20-11-19-14/h3,6-8,11H,2,4,9-10H2,1H3,(H,18,19,20)
|
|
| 化学名 |
|
|
| 别名 |
|
|
| HS Tariff Code |
2934.99.9001
|
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
|
|||
|---|---|---|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 2.5 mg/mL (6.73 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 25.0 mg/mL澄清DMSO储备液加入到400 μL PEG300中,混匀;然后向上述溶液中加入50 μL Tween-80,混匀;加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: ≥ 2.5 mg/mL (6.73 mM) (饱和度未知) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 25.0 mg/mL澄清DMSO储备液加入900 μL 20% SBE-β-CD生理盐水溶液中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 View More
配方 3 中的溶解度: ≥ 2.5 mg/mL (6.73 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 配方 4 中的溶解度: 0.5% CMC+0.25% Tween 80:30mg/mL 配方 5 中的溶解度: 2.5 mg/mL (6.73 mM) in 0.5% Methylcellulose/saline water (这些助溶剂从左到右依次添加,逐一添加), 悬浊液; 超声助溶。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.6924 mL | 13.4618 mL | 26.9237 mL | |
| 5 mM | 0.5385 mL | 2.6924 mL | 5.3847 mL | |
| 10 mM | 0.2692 mL | 1.3462 mL | 2.6924 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
| NCT Number | Recruitment | interventions | Conditions | Sponsor/Collaborators | Start Date | Phases |
| NCT04901325 | Recruiting | Drug: Baricitinib | RPyoderma Gangrenosum Skin Diseases |
Oregon Health and Science University | October 2023 | Phase 2 |
| NCT05852171 | Recruiting | Drug: Baricitinib | Mastitis Chronic Idiopathic Granulomatous Mastitis |
First Affiliated Hospital of Zhejiang University |
January 1, 2023 | Phase 2 |
| NCT05074420 | Recruiting | Drug: Baricitinib | Covid19 Corona Virus Infection |
Eli Lilly and Company | December 21, 2021 | Phase 3 |
| NCT06240351 | Not yet recruiting | Drug: Baricitinib 4 MG Oral Tablet | Frontal Fibrosing Alopecia | University of Alabama at Birmingham |
June 1, 2024 | Phase 4 |
Cellular activity of INCB028050.J Immunol.2010 May 1;184(9):5298-307. |
Anti-inflammatory and DMARD activity of once daily INCB028050 in rats with established disease in the adjuvant arthritis model.J Immunol.2010 May 1;184(9):5298-307. |
Suppression of delayed-type hypersensitivity by INCB028050.J Immunol.2010 May 1;184(9):5298-307. |
INCB028050 is efficacious and well tolerated independently of effects on humoral immunity.J Immunol.2010 May 1;184(9):5298-307. |
INCB028050 improves clinical and histologic signs of disease in the murine CIA model. |
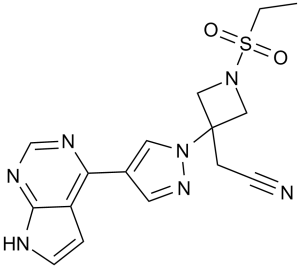
 Frevecitinibum
Frevecitinibum
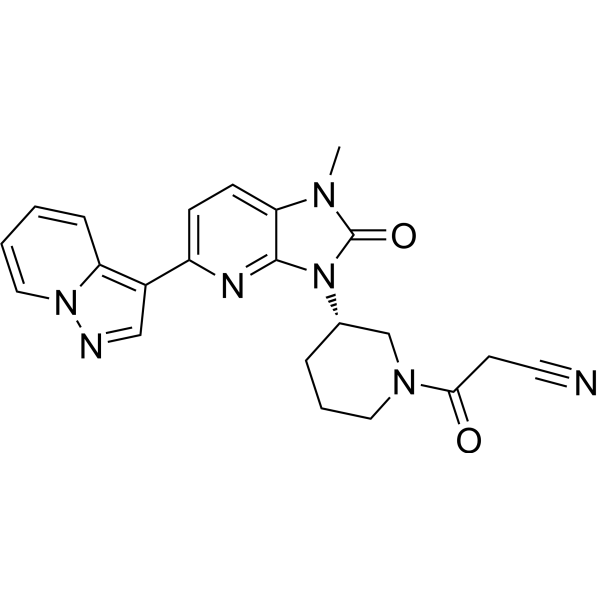
 Girocitinibum
Girocitinibum
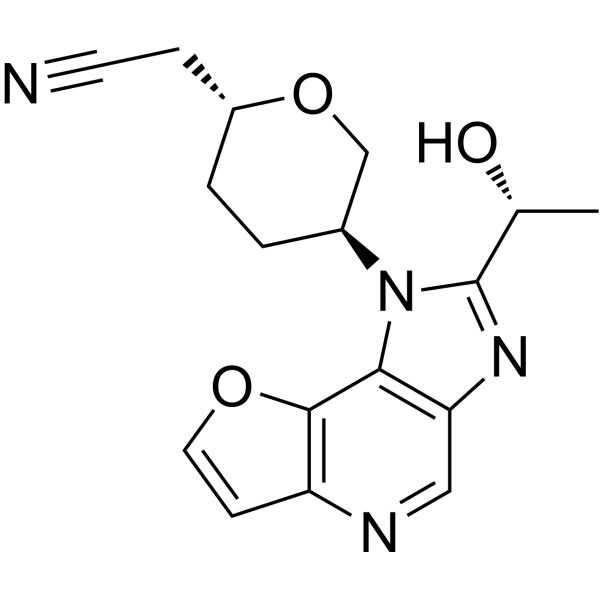
 Envudeucitinibum
Envudeucitinibum
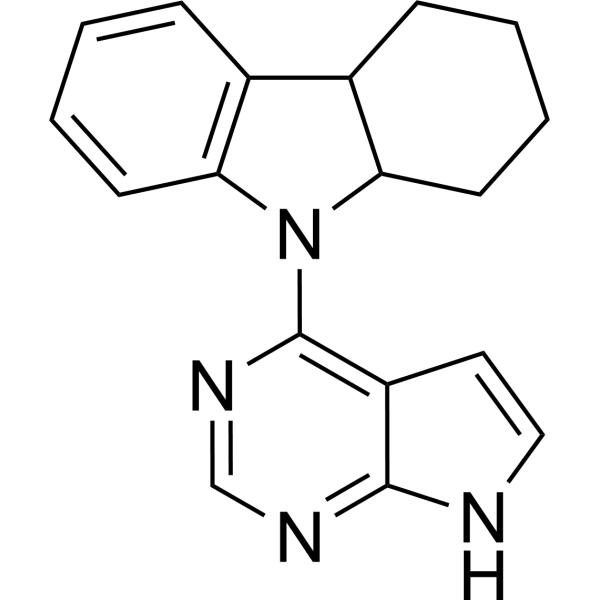 CP-352664
CP-352664
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1






























 COA
COA
")
")
")
")
")
