| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 10 mM * 1 mL in DMSO |
|
||
| 5mg |
|
||
| 10mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| 500mg |
|
||
| 1g |
|
||
| 2g |
|
||
| Other Sizes |
|

| 靶点 |
PPARα (EC50 = 0.63 μM); PPARγ (EC50 = 32 μM)
|
||
|---|---|---|---|
| 体外研究 (In Vitro) |
作为 PPARα 的激动剂,pirinixic Acid (Wy-14643) 对小鼠 PPARα 和 PPARγ 的 EC50 值为 0.63 μM 和 32 μM,对人体 PPARα 和 PPARγ 的 EC50 分别为 5.0 μM、60 μM、35 μM 和 PPARδ [1]。在滑膜成纤维细胞中,pirinixic Acid(Wy-14643;0、10、100 μM)可增加 PPAR-α 蛋白的表达。 LPS 刺激的滑膜成纤维细胞生成 NO 和 PGE2 被吡尼尼酸 (0、10、100 μM) 抑制。此外,pirinixic Acid 可有效抑制滑膜成纤维细胞中 LPS 诱导的 NF-kB 激活、IkB 磷酸化和 TF,并下调这些细胞中炎症介质的产生,包括 VCAM-1、ICAM-1、ET-1 和 TF 。 PPAR-α 使细胞沉默,而吡尼尼酸对 NF-kB 核转位几乎没有影响 [2]。
|
||
| 体内研究 (In Vivo) |
在肥胖大鼠中,pirinixic Acid(Wy-14643;10 mg/kg,IV)可降低 MDA 水平和肝损伤。在假手术组和缺血再灌注(IR)组中,吡尼尼酸同样增加了 SIRT1 活性,但对 SIRT3 蛋白的产生没有影响。在大鼠中,pirinixic 酸可以预防内质网应激 (ERS) 并提高 NAD+ 和 ATP 水平 [3]。
|
||
| 细胞实验 |
WY-14643是一种有效的过氧化物酶体增殖物激活受体-α (PPAR-α)激动剂,已被描述为有益于调节许多哺乳动物细胞的炎症。在这里,我们研究WY-14643对脂多糖(LPS)诱导的滑膜成纤维细胞的潜在抗炎作用。WY-14643显著抑制LPS诱导的NO和PGE2的生成。此外,WY-14643显著抑制细胞内黏附分子-1 (ICAM-1)、血管细胞黏附分子-1 (VCAM-1)、内皮素-1 (ET-1)、组织因子(TF) mRNA表达,抑制促炎因子白介素-6 (IL-6)、IL-1β、肿瘤坏死因子-α (TNF-α)、单核细胞趋化蛋白-1 (MCP-1)的分泌。此外,WY-14643显著降低NF-kB的转录活性和核易位,增强IkB的磷酸化,表明WY-14643的抗炎作用是通过NF-kB依赖途径介导的。WY-14643在PPAR-α沉默细胞中未能发挥其抗炎功能,提示PPAR-α的作用。这些发现可能有助于进一步研究PPAR-α药理激活在OA临床治疗中的转化[2]。
|
||
| 动物实验 |
|
||
| 毒性/毒理 (Toxicokinetics/TK) |
Interactions
Inflammatory mediators orchestrate the host immune and metabolic response to acute bacterial infections and mediate the events leading to septic shock. Tumor necrosis factor (TNF) has long been identified as one of the proximal mediators of endotoxin action. Recent studies have implicated peroxisome proliferator-activated receptor alpha (PPARalpha) as a potential target to modulate regulation of the immune response. Since PPARalpha activators, which are hypolipidemic drugs, are being prescribed for a significant population of older patients, it is important to determine the impact of these drugs on the host response to acute inflammation. Therefore, we examined the role of PPARalpha activators on the regulation of TNF expression in a mouse model of endotoxemia. CD-1 mice treated with dietary fenofibrate or Wy-14,643 had fivefold-higher lipopolysaccharide (LPS)-induced TNF plasma levels than LPS-treated control-fed animals. Higher LPS-induced TNF levels in drug-fed animals were reflected physiologically in significantly lower glucose levels in plasma and a significantly lower 50% lethal dose than those in LPS-treated control-fed animals. Utilizing PPARalpha wild-type (WT) and knockout (KO) mice, we showed that the effect of fenofibrate on LPS-induced TNF expression was indeed mediated by PPARalpha. PPARalpha WT mice fed fenofibrate also had a fivefold increase in LPS-induced TNF levels in plasma compared to control-fed animals. However, LPS-induced TNF levels were significantly decreased and glucose levels in plasma were significantly increased in PPARalpha KO mice fed fenofibrate compared to those in control-fed animals. Data from peritoneal macrophage studies indicate that Wy-14,643 modestly decreased TNF expression in vitro. Similarly, overexpression of PPARalpha in 293T cells decreased activity of a human TNF promoter-luciferase construct. The results from these studies suggest that any anti-inflammatory activity of PPARalpha in vivo can be masked by other systemic effects of PPARalpha activators. Non-Human Toxicity Values LD50 Rat oral 4150 mg/kg LD50 Mouse oral 1600 mg/kg |
||
| 参考文献 | |||
| 其他信息 |
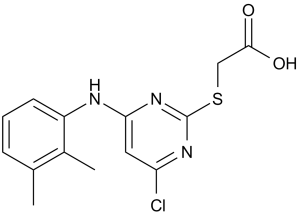
Pirinixic acid is a member of pyrimidines, an organochlorine compound and an aryl sulfide. It is functionally related to an acetic acid.
Pirinixic Acid is a synthetic thiacetic acid derivative used in biomedical research, carcinogenic Pirinixic acid is a peroxisome proliferator that activates specific peroxisome proliferator-activated receptors (PPAR). PPARs play an important role in diverse cellular functions, including lipid metabolism, cell proliferation, differentiation, adipogenesis, and inflammatory signaling. (NCI04) Mechanism of Action Effects of several classes of peroxisomal proliferators on peroxisomal functions, hepatomegaly, hepatocarcinogenesis and lipid metabolism have been extensively investigated in rodents. Less is known about influences of these agents, some used as hypolipidemic drugs, on various metabolic parameters in humans. We examined effects of clofibrate, di(2-ethyl-hexyl)phthalate (DEHP) and pirinixic acid (WY-14,643) on phospholipid metabolism in human fibroblasts in culture. Clofibrate inhibited incorporation of [1-(14)C]hexadecanol and [1-(14)C]linolenic acid into ethanolamine phosphoglycerides in a time- and concentration-dependent manner; labeling of plasmalogens and non-plasmalogen ethanolamine phosphoglycerides was reduced by 40-80% compared to a generalized 10-30% inhibition of labeling of other phospholipids, including phosphatidylcholine. In pulse and pulse-chase experiments, selective inhibition of incorporation of [1,2-(14)C]ethanolamine, compared to [methyl-(3)H]choline, confirmed relative specificity of inhibition of ethanolamine phosphoglycerides. Similar concentration dependence and specificity for inhibition of phospholipid turnover was observed for DEHP and WY-14,643, in both control and mutant (Zellweger and adrenoleukodystrophy) fibroblasts, in the absence of major effects on peroxisomal markers. These observations that peroxisomal proliferators specifically inhibit ethanolamine phosphoglyceride turnover in human fibroblasts should be considered when assessing the efficacy and safety of such agents as hypolipidemic drugs or when evaluating mechanisms of proliferator action at the cellular level. Pirinixic acid (Wy-14,643) is an agonist of the peroxisome proliferator-activated receptor (PPAR) subtype alpha exhibiting beneficial effects in various inflammation-related processes in a slow, long-termed fashion. We recently showed that alpha-substituted pirinixic acid derivatives are agonists of PPAR alpha and act as dual inhibitors of 5-lipoxygenase (5-LO, EC 1.13.11.34) and the microsomal prostaglandin E(2) synthase-1 (EC 5.3.99.3). Here, we explored short-term effects of alpha-substituted pirinixic acid derivatives on typical neutrophil functions evoked by the agonist N-formyl-methionyl-leucyl-phenylalanine (fMLP) including leukotriene formation, generation of reactive oxygen species, and release of human leukocyte elastase (EC 3.4.21.37), and we investigated the modulation of related signalling pathways. Pirinixic acid derivatives that are substituted with alkyl residues in alpha-position of the carboxylic group and with a 6-aminoquinoline residue at the pyrimidine moiety cause inhibition of leukotriene formation, reactive oxygen species formation, and leukocyte elastase release in response to fMLP. In parallel, Ca(2+) mobilisation and the phosphorylation (activation) of p38 mitogen-activated protein kinase was significantly reduced, whereas phosphorylation of the extracellular signal-regulated kinase-2 was unaffected. Pirinixic acid itself was not or only marginally active in all these assays. Conclusively, targeted structural modification of pirinixic acid leads to bioactive compounds that display immediate anti-inflammatory properties in human neutrophils with potential therapeutic value. Normal function of the peroxisome proliferator-activated receptor alpha (PPARalpha) is crucial for the regulation of hepatic fatty acid metabolism. Fatty acids serve as ligands for PPARalpha, and when fatty acid levels increase, activation of PPARalpha induces a battery of fatty acid-metabolizing enzymes to restore fatty acid levels to normal. Hepatic fatty acid levels are increased during ethanol consumption. However, results of in vitro work showed that ethanol metabolism inhibited the ability of PPARalpha to bind DNA and activate reporter genes. This observation has been further studied in mice. Four weeks of ethanol feeding of C57BL/6J mice also impairs fatty acid catabolism in liver by blocking PPARalpha-mediated responses. Ethanol feeding decreased the level of retinoid X receptor alpha (RXRalpha) as well as the ability of PPARalpha/RXR in liver nuclear extracts to bind its consensus sequence, and the levels of mRNAs for several PPARalpha-regulated genes were reduced [long-chain acyl coenzyme A (acyl-CoA) dehydrogenase and medium-chain acyl-CoA dehydrogenase] or failed to be induced (acyl-CoA dehydrogenase, liver carnitine palmitoyl-CoA transferase I, very long-chain acyl-CoA synthetase, very long-chain acyl-CoA dehydrogenase) in livers of the ethanol-fed animals. Consistent with this finding, ethanol feeding did not induce the rate of fatty acid beta-oxidation, as assayed in liver homogenates. Inclusion of WY14,643, a PPARalpha agonist, in the diet restored the DNA-binding activity of PPARalpha/RXR, induced mRNA levels of several PPARalpha target genes, stimulated the rate of fatty acid beta-oxidation in liver homogenates, and prevented fatty liver in ethanol-fed animals. Blockade of PPARalpha function during ethanol consumption contributes to the development of alcoholic fatty liver, which can be overcome by WY14,643. Endothelium injury is a primary event in atherogenesis, which is followed by monocyte infiltration, macrophage differentiation, and smooth muscle cell migration. Peroxisome proliferator-activated receptors (PPARs) are transcription factors now recognized as important mediators in the inflammatory response. The aim of this study was to develop a human endothelial model to evaluate anti-inflammatory properties of PPAR activators. PPAR proteins (alpha, delta and gamma) are expressed in EAhy926 endothelial cells (ECs). Pirinixic acid (Wy-14643), fenofibrate, fenofibric acid, the Merck ligand PPARdelta activator L-165041, 15-deoxy-Delta(12,14)-prostaglandin J2, but not rosiglitazone (BRL-49653) inhibited the induced expression of vascular cell adhesion molecule-1 (VCAM-1), as measured by enzyme linked immunosorbent assay (ELISA), and monocyte binding to activated-EAhy926 cells. The PPARdelta activator L-165041 had the greatest potency to reduce cytokine-induced monocyte chemotactic protein-1 (MCP-1) secretion. All PPAR activators tested which impaired VCAM-1 expression reduced significantly nuclear p65 amount. These results show that EAhy926 endothelial cells are an adequate tool to substantiate and characterize inflammatory impacts of PPAR activators. For more Mechanism of Action (Complete) data for Pirinixic acid (10 total), please visit the HSDB record page. |
| 分子式 |
C14H14CLN3O2S
|
|
|---|---|---|
| 分子量 |
323.8
|
|
| 精确质量 |
323.049
|
|
| 元素分析 |
C, 51.93; H, 4.36; Cl, 10.95; N, 12.98; O, 9.88; S, 9.90
|
|
| CAS号 |
50892-23-4
|
|
| 相关CAS号 |
|
|
| PubChem CID |
5694
|
|
| 外观&性状 |
Typically exists as White to off-white solids at room temperature
|
|
| 密度 |
1.4±0.1 g/cm3
|
|
| 沸点 |
514.4±50.0 °C at 760 mmHg
|
|
| 熔点 |
155°C
|
|
| 闪点 |
264.9±30.1 °C
|
|
| 蒸汽压 |
0.0±1.4 mmHg at 25°C
|
|
| 折射率 |
1.658
|
|
| LogP |
4.92
|
|
| tPSA |
100.41
|
|
| 氢键供体(HBD)数目 |
2
|
|
| 氢键受体(HBA)数目 |
6
|
|
| 可旋转键数目(RBC) |
5
|
|
| 重原子数目 |
21
|
|
| 分子复杂度/Complexity |
361
|
|
| 定义原子立体中心数目 |
0
|
|
| SMILES |
ClC1C([H])=C(N=C(N=1)SC([H])([H])C(=O)O[H])N([H])C1=C([H])C([H])=C([H])C(C([H])([H])[H])=C1C([H])([H])[H]
|
|
| InChi Key |
SZRPDCCEHVWOJX-UHFFFAOYSA-N
|
|
| InChi Code |
InChI=1S/C14H14ClN3O2S/c1-8-4-3-5-10(9(8)2)16-12-6-11(15)17-14(18-12)21-7-13(19)20/h3-6H,7H2,1-2H3,(H,19,20)(H,16,17,18)
|
|
| 化学名 |
[[4-chloro-6-[(2,3-dimethylphenyl)amino]-2-pyrimidinyl]thio]-acetic acid
|
|
| 别名 |
|
|
| HS Tariff Code |
2934.99.9001
|
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
|
|||
|---|---|---|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 2.08 mg/mL (6.42 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 20.8 mg/mL澄清DMSO储备液加入400 μL PEG300中,混匀;然后向上述溶液中加入50 μL Tween-80,混匀;加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: ≥ 2.08 mg/mL (6.42 mM) (饱和度未知) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 20.8 mg/mL澄清DMSO储备液加入900 μL 20% SBE-β-CD生理盐水溶液中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 View More
配方 3 中的溶解度: ≥ 2.08 mg/mL (6.42 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 3.0883 mL | 15.4416 mL | 30.8833 mL | |
| 5 mM | 0.6177 mL | 3.0883 mL | 6.1767 mL | |
| 10 mM | 0.3088 mL | 1.5442 mL | 3.0883 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
|
|---|
|
|
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1





























 COA
COA
")
")
")




