| 规格 | 价格 | |
|---|---|---|
| 500mg | ||
| 1g | ||
| Other Sizes |
| 靶点 |
VEGFR2 (IC50 = 40 nM); VEGFR3 (IC50 = 110 nM); EGFR/HER1 (IC50 = 500 nM)
|
|
|---|---|---|
| 体外研究 (In Vitro) |
体外活性:Vandetanib 还抑制 VEGFR3 和 EGFR,IC50 分别为 110 nM 和 500 nM。 Vandetanib对PDGFRβ、Flt1、Tie-2和FGFR1不敏感,IC50为1.1-3.6 μM,而对MEK、CDK2、c-Kit、erbB2、FAK、PDK1、Akt和IGF-1R几乎没有活性,IC50以上10μM。 Vandetanib 抑制 VEGF、EGF 和 bFGF 刺激的 HUVEC 增殖,IC50 分别为 60 nM、170 nM 和 800 nM,对基底内皮细胞生长没有影响。 Vandetanib 抑制肿瘤细胞生长,IC50 为 2.7 μM (A549) 至 13.5 μM (Calu-6)。 Vandetanib 对基础 ABCG2-ATP 酶显示出抑制作用。亲代和表达 ABCG2 的 A431 细胞对凡德他尼表现出相似的敏感性。暴露于 EGFR 抑制剂会降低 A431 细胞中的 pEGFR 水平,而凡德他尼仅表现出中等效果。 Vandetanib 显示出轻微但可测量的效果,而吉非替尼、佩利替尼和来那替尼完全抑制 ABCG2 介导的米托蒽醌从 A431/ABCG2 细胞中的流出,类似于特定的 ABCG2 抑制剂 Ko143。 Vandetanib 抑制 PC3wt 和 PC3R 细胞系,IC50 相似,分别为 13.3 μM 和 11.5 μM。 Vandetanib 抑制 HUVEC 中 VEGFR2 和肝癌细胞中 EGFR 的磷酸化并抑制细胞增殖。凡德他尼引起 GEO 和 OVCAR-3 细胞中 G0-G1 期细胞的积累,并增加 OVCAR-3、ZR-75-1、MCF-10A ras 和 GEO 细胞的凋亡。 Vandetanib 在小鼠 NIH-EGFR 成纤维细胞和人 MCF-10A ras 乳腺癌细胞(两种过度表达人 EGFR 的细胞系)中对 EGFR 磷酸化产生剂量依赖性抑制。 Vandetanib 治疗可对 7 种具有功能性 EGFR 但缺乏 VEGFR2 的人类细胞系(乳腺、结肠、胃和卵巢)中的软琼脂生长产生剂量依赖性抑制。激酶测定:凡德他尼与酶、10 mM MnCl2 和 2 μM ATP 在涂有聚(Glu、Ala、Tyr)6:3:1 无规共聚物底物的 96 孔板中孵育。然后通过与小鼠 IgG 抗磷酸酪氨酸 4G10 抗体、辣根过氧化物酶连接的绵羊抗小鼠免疫球蛋白抗体和 2,2'-azino-bis(3-乙基苯并噻唑啉-6-磺酸) 连续孵育来检测磷酸化酪氨酸。该方法适用于检查与 EGFR、PDGFRβ、Tie-2、FGFR1、c-kit、erbB2、IGF-1R 和 FAK 相关的酪氨酸激酶的选择性。所有酶测定(酪氨酸或丝氨酸-苏氨酸)均使用等于或略低于各自 Km (0.2–14 μM) 的适当 ATP 浓度。在 96 孔板中使用相关闪烁邻近分析 (SPA) 检查相对于丝氨酸-苏氨酸激酶(CDK2、AKT 和 PDK1)的选择性。 CDK2 检测包含 10 mM MnCl2、4.5 μM ATP、0.15 μCi 的 [γ-33 P]ATP/反应、50 mM HEPES (pH 7.5)、1 mM DTT、0.1 mM 原钒酸钠、0.1 mM 氟化钠、10 mM 甘油磷酸钠、1 mg/mL BSA 组分 V 和视网膜母细胞瘤底物(视网膜母细胞瘤基因的一部分,792-928,在谷胱甘肽 S-转移酶表达系统中表达;最终浓度为 0.22 μM)。反应在室温下进行 60 分钟,然后用 150 μL 含有 EDTA(62 mM 最终浓度)、3 μg 兔免疫球蛋白抗谷胱甘肽 S-转移酶抗体和蛋白 A SPA-聚乙烯甲苯的溶液猝灭 2 小时珠子(0.8 毫克/反应)。然后将板密封、离心(1200×g,5分钟),并在微孔板闪烁计数器上计数30秒。细胞测定:将肿瘤细胞(Calu-6、PC-3、MDA-MA-231、SKOV-3、SW620、A549、A431、B16-F10(AP3) 和 Lewis 肺细胞)以预定密度接种在各自的培养基中已知在检测期间能够实现对数细胞生长(PC-3,500 个细胞/孔;所有其他,1000 个细胞/孔)。将板孵育 24 小时(37 °C,CO2),然后添加 Vandetanib (0.1–100 μM) 或媒介物(培养基中的 0.1% DMSO)。将板再孵育 72 小时,然后通过 β 计数器通过 [3 H]胸苷掺入评估细胞增殖。
|
|
| 体内研究 (In Vivo) |
Vandetanib(2.5 mg/kg,静脉注射)可将 VEGF 诱导的低血压逆转 63%,但不会显着影响 bFGF 诱导的低血压。 Vandetanib (100 mg/kg) 抑制肿瘤诱导的血管形成 79%。 Vandetanib(12.5-100 mg/kg,口服)在人类肿瘤异种移植物中显示出良好的肿瘤生长抑制作用,包括 Calu-6、PC-3、MDA-MA-231、SKOV-3、SW620、A549、A431、B16-F10(AP3) )和 Lewis Lung,对体重影响很小。在 PC3wt 异种移植物中,单独给予凡德他尼会发挥矛盾的肿瘤生长刺激作用。在PC3R异种移植物中,低剂量Vandetanib(25 mg/kg)相对于对照没有显着效果,而高剂量(50 mg/kg)与对照相比显着抑制肿瘤生长。相比之下,高剂量组合显示 PC3R 细胞中凡德他尼 50 mg/kg 和多西他赛 30 mg/kg 之间存在显着的负相互作用。在荷瘤小鼠中,Vandetanib抑制肿瘤组织中VEGFR2和EGFR的磷酸化,显着降低肿瘤血管密度,增强肿瘤细胞凋亡,抑制肿瘤生长,提高生存率,减少肝内转移数量,并上调VEGF、TGF-α和肿瘤组织中的EGF。 Vandetanib 治疗不会导致严重不良事件,包括 ALT 异常、骨髓抑制或体重减轻。 Vandetanib 治疗携带可触及 GEO 结肠癌异种移植物(对 EGFR 信号传导抑制敏感)的裸鼠,可诱导剂量依赖性肿瘤生长抑制
|
|
| 酶活实验 |
在涂有聚(Glu、Ala、Tyr)6:3:1 无规共聚物底物的 96 孔板中,将凡德他尼与酶、10 mM MnCl2 和 2 μM ATP 一起孵育。下一步是通过依次孵育 2,2'-azino-bis(3-乙基苯并噻唑啉-6-磺酸)、辣根过氧化物酶连接的绵羊抗小鼠免疫球蛋白抗体和小鼠 IgG 抗磷酸酪氨酸 4G10 抗体来鉴定磷酸化酪氨酸。为了研究对与 FGFR1、c-kit、erbB2、IGF-1R、FAK、PDGFRβ、Tie-2 和 FGFR1 相关的酪氨酸激酶的选择性,对该方法进行了修改。所有酶测定(酪氨酸或丝氨酸-苏氨酸)均使用等于或略低于相应 Km (0.2–14 μM) 的适当 ATP 浓度。使用相关闪烁邻近分析 (SPA) 在 96 孔板中研究针对丝氨酸-苏氨酸激酶(CDK2、AKT 和 PDK1)的选择性。 CDK2 检测的条件如下:10 mM MnCl2、4.5 μM ATP、0.15 μCi [γ-33 P]ATP/反应、50 mM HEPES( pH 7.5)、1 mM DTT、0.1 mM 原钒酸钠、0.1 mM 氟化钠、10 mM 甘油磷酸钠、1 mg/mL BSA 组分 V 和视网膜母细胞瘤底物(视网膜母细胞瘤基因的一部分,792-928,以谷胱甘肽 S-转移酶表达系统;0.22 μM 初始浓度)。反应在室温下进行 60 分钟,然后使用 150 μL 溶液淬灭 2 小时,该溶液含有 0.8 mg/反应的 Protein A SPA-聚乙烯甲苯珠、3 μg 兔免疫球蛋白抗谷胱甘肽 S-转移酶抗体和EDTA(62 mM 最终浓度)。之后,将板密封,在1200 xg下离心五分钟,并使用微板闪烁计数器计数三十秒。
|
|
| 细胞实验 |
MTT 测定经过修改以测量生长抑制。简而言之,细胞以每孔 2000 个细胞的密度接种在 96 孔板中后,暴露于凡德他尼或吉非替尼 72 小时。每个测定进行三次。对于每种药物,50% 抑制浓度 (IC50) 使用平均值±标准差 (SD) 计算。
|
|
| 动物实验 |
|
|
| 药代性质 (ADME/PK) |
Absorption, Distribution and Excretion
Slow- peak plasma concentrations reached at a median 6 hours. On multiple dosing, Vandetanib accumulates about 8 fold with steady state reached after around 3 months. About 69% was recovered following 21 days after a single dose of vandentanib. 44% was found in feces and 25% in urine. Vd of about 7450 L. Vandetanib binds to human serum albumin and a1-acid-glycoprotein with in vitro protein binding being approximately 90%. In ex vivo plasma samples from colorectal cancer patients at steady state exposure after 300 mg once daily, the mean percentage protein binding was 94%. Within a 21-day collection period after a single dose of (14)C-vandetanib, approximately 69% was recovered with 44% in feces and 25% in urine. Excretion of the dose was slow and further excretion beyond 21 days would be expected based on the plasma half-life. Vandetanib was not a substrate of hOCT2 expressed in HEK293 cells. Vandetanib inhibits the uptake of the selective OCT2 marker substrate 14C-creatinine by HEK-OCT2 cells, with a mean IC50 of 2.1 ug/mL. This is higher than vandetanib plasma concentrations (0.81 ug/mL) observed after multiple dosing at 300 mg. Inhibition of renal excretion of creatinine by vandetanib provides an explanation for increases in plasma creatinine seen in human subjects receiving vandetanib. Following oral administration of Caprelsa, absorption is slow with peak plasma concentrations typically achieved at a median of 6 hours, range 4-10 hours, after dosing. Vandetanib accumulates approximately 8-fold on multiple dosing with steady state achieved in approximately 3 months. Exposure to vandetanib is unaffected by food. The protein binding of (14)C-Vandetanib in plasma of mice, rats, rabbits dogs and human was moderate, from 83 to 90%. The tissue distribution of vandetanib and/or metabolites in pigmented and non pigmented male rats after single oral dosing was slow but extensive, and consistent with the distribution pattern of a lipophilic compound. Highest concentrations of vandetanib and/or its metabolites were seen in the majority of tissues at 6-8 hours after administration. The distribution of radioactivity to brain was evident. Retention of radioactivity was seen in pigmented tissues indicating melanin affinity. A significant distribution of radioactivity was seen in milk of lactating rats and further on in the plasma of suckling pups. For more Absorption, Distribution and Excretion (Complete) data for Vandetanib (8 total), please visit the HSDB record page. Metabolism / Metabolites Unchanged vandentanib and metabolites vandetanib N-oxide and N-desmethyl vandetanib were detected in plasma, urine and feces. N-desmethyl-vandetanib is primarily produced by CYP3A4, and vandetanib-N-oxide is primarily produced by flavin–containing monooxygenase enzymes FMO1 and FMO3. The metabolism of vandetanib seemed to be similar in the toxicology species, rat and dog, as well as in mouse and human. The 2 major metabolites identified in excreta, were N-desmethyl-vandetanib and vandetanib-N-oxide. In mouse, a minor metabolite was also identified as O-desalkyl-vandetanib glucuronid. A glucuronide conjugate was also detected in human urine. Metabolism as well as biliary excretion appears to be most important for the elimination of vandetanib in preclinical species. CYP identification studies in vitro, suggest that CYP3A4 is involved in the formation of N-desmethyl-Vandetanib. vandetanib-N-oxide is formed via FMO1 and FMO3 (FMO=flavine mono-oxygenase). Both these enzymes are also found in kidney indicating that renal excretion might be contributed to the clearance of vandetanib. Following oral dosing of (14)C-vandetanib, unchanged vandetanib and metabolites vandetanib N-oxide and N-desmethyl vandetanib were detected in plasma, urine and feces. A glucuronide conjugate was seen as a minor metabolite in excreta only. N-desmethyl-vandetanib is primarily produced by CYP3A4 and vandetanib-N-oxide by flavin-containing monooxygenase enzymes FMO1 and FMO3. N-desmethyl-vandetanib and vandetanib-N-oxide circulate at concentrations of approximately 7-17% and 1.4-2.2%, respectively, of those of vandetanib. ... In plasma, concentrations of total radioactivity were higher than vandetanib concentrations at all time points, indicating the presence of circulating metabolites. Unchanged vandetanib and 2 anticipated metabolites (N-desmethylvandetanib and vandetanib N-oxide) were detected in plasma, urine, and feces. A further trace minor metabolite (glucuronide conjugate) was found in urine and feces. ... Unchanged vandetanib and N-desmethyl and N-oxide metabolites were detected in plasma, urine, and feces. Biological Half-Life Median half life of 19 days. ... Caprelsa at the 300 mg dose in medullary thyroid cancer (MTC) patients /is/ characterized by a ... median plasma half-life of 19 days. ... Vandetanib was absorbed and eliminated slowly with a half life of approximately 10 days after single oral doses. ... |
|
| 毒性/毒理 (Toxicokinetics/TK) |
Toxicity Summary
IDENTIFICATION AND USE: Vandetanib is a white to off white powder that is formulated into film-coated tablets. Vandetanib is a multitargeted tyrosine kinase inhibitor used for the treatment of symptomatic or progressive medullary thyroid cancer in patients with unresectable locally advanced or metastatic disease. Because of the risk of QT prolongation, torsades de pointes, and sudden death, the US Food and Drug Administration (FDA) requires a Risk Evaluation and Mitigation Strategy (REMS) for vandetanib. Under the terms of the REMS program, vandetanib is available only under a restricted distribution program. It was granted orphan drug status by the FDA. HUMAN EXPOSURE AND TOXICITY: Vandetanib prolongs the QT interval in a concentration-dependent manner. Torsades de pointes (a distinctive polymorphic ventricular tachycardia in which the QRS amplitude varies and the QRS complexes appear to twist around), ventricular tachycardia, and sudden death have all been reported in patients receiving vandetanib. Vandetanib should not be used in patients who have a history of torsades de pointes, congenital long QT syndrome, bradyarrhythmias, or uncompensated heart failure, or in patients with electrolyte disturbances. Hypocalcemia, hypokalemia, and/or hypomagnesemia must be corrected prior to the administration of vandetanib. Other toxicities that are associated with the use of vandetanib and have resulted in fatalities include: severe skin reactions (including Stevens-Johnson syndrome), interstitial lung disease or pneumonitis, ischemic cerebrovascular events, serious hemorrhagic events and heart failure. Vandetanib may also cause fetal harm if administered to pregnant women. Pregnancy should therefore be avoided during vandetanib therapy. Vandetanib was not clastogenic to cultured human lymphocytes. ANIMAL STUDIES: In the rat, a single oral dose at 2000 mg/kg was not tolerated and all animals died or were killed for humane reasons by Day 4. Histopathological findings in these rats included hepatocyte vacuolation, fat deposition and necrosis in the liver, ulceration in the stomach, mucosal single cell necrosis and erosion in the duodenum, and macrophage vacuolation in the spleen. There were no adverse effects in rats dosed at 1000 mg/kg. A single oral dose of vandetanib at 2000 mg/kg to mice was not tolerated and all animals died or were killed for humane reasons on Day 1. A single oral dose of 1000 mg/kg resulted in the death of 1 out of 10 mice. There were no salient histopathology findings except for ulceration in the stomach in 1 animal dosed at 2000 mg/kg. In 1, 6 and 9 month studies, the dose limiting toxicities included gastrointestinal effects in dogs (including loose/abnormal feces, emesis and body weight loss), and skin toxicity and hepatotoxicity in rats. Vandetanib had no effect on copulation or fertility in male rats, while in female rats there was a trend towards increased estrus cycle irregularity, a slight reduction in pregnancy and an increase in post-implantation loss. In rats, vandetanib demonstrated the potential to cause embryo-fetal loss, delayed fetal development, heart vessel abnormalities and precocious ossification of some skull bones. In a rat pre- and post-natal development study, at doses producing maternal toxicity during gestation and/or lactation, vandetanib increased pre-birth loss and reduced post-natal pup growth. Vandetanib showed no mutagenic potential in 4 strains of Salmonella typhimurium (TA1535, TA1537, TA98 and TA100) and 2 strains of Escherichia coli (WP2P and WP2 uvrA) with or without metabolic activation. Hepatotoxicity In large clinical trials of vandetanib, abnormalities in routine liver tests were common with serum aminotransferase elevations, occurring in up to half of patients and rising above 5 times the upper limit of normal (ULN) 2% to 5% of patients. In prelicensure trials of vandetanib in thyroid cancer, there were no reports of clinically apparent liver injury with jaundice or hepatic failure. Since approval and more wide scale use, there have been no published reports of hepatotoxicity due to vandetanib and the product label does not include discussion of hepatotoxicity. However, many of the kinase inhibitors used in cancer chemotherapy have been implicated in cases of clinically apparent liver injury which typically arises within the first 2 to 12 weeks of therapy, presenting with symptoms of fatigue, nausea and jaundice and a hepatocellular pattern of serum enzyme elevations without immunoallergic or autoimmune features. Several tyrosine kinase inhibitors (imatinib, nilotinib) have also been implicated in causing reactivation of hepatitis B. Likelihood score: E* (unproven but suspected rare cause of clinically apparent liver injury). Effects During Pregnancy and Lactation ◉ Summary of Use during Lactation No information is available on the clinical use of vandetanib during breastfeeding. Because vandetanib is 90% bound to plasma proteins, the amount in milk is likely to be low. However, its half-life is 19 days and it might accumulate in the infant. The manufacturer recommends that breastfeeding be discontinued during vandetanib therapy and for 4 months after the last dose. ◉ Effects in Breastfed Infants Relevant published information was not found as of the revision date. ◉ Effects on Lactation and Breastmilk Relevant published information was not found as of the revision date. Protein Binding Protein binding of about 90%. Interactions Concomitant use of vandetanib with drugs known to prolong the QT interval, including class Ia (e.g., disopyramide, procainamide, quinidine) and class III (e.g., amiodarone, sotalol, dofetilide) antiarrhythmic agents, some anti-infectives (e.g., clarithromycin, gatifloxacin, moxifloxacin), some antipsychotic agents (e.g., chlorpromazine, thioridazine, haloperidol, asenapine, olanzapine, paliperidone, pimozide, quetiapine, ziprasidone), some type 3 serotonin (5-HT3) receptor antagonists used as antiemetic agents (e.g., dolasetron, granisetron, ondansetron), chloroquine, methadone, and tetrabenazine should be avoided. If a drug known to prolong the QT interval must be administered, more frequent ECG monitoring is recommended. If a 5-HT3 receptor antagonist is clinically necessary, some clinicians prefer granisetron because its effects on ECG intervals are less pronounced than those observed with dolasetron or ondansetron. Inducers of CYP3A4 can alter plasma vandetanib concentrations. Concomitant use of vandetanib with potent CYP3A4 inducers (e.g., carbamazepine, dexamethasone, phenobarbital, phenytoin, rifabutin, rifampin, rifapentine) should be avoided. St. John's wort (Hypericum perforatum) may unpredictably decrease vandetanib exposure, and concomitant use of vandetanib with this agent also should be avoided. Caprelsa increased plasma concentrations of digoxin. Use caution and closely monitor for toxicities when administering Caprelsa with digoxin. Capresla increased plasma concentrations of metformin that is transported by the organic cation transporter type 2 (OCT2). Use caution and closely monitor for toxicities when administering Capresla with drugs that are transported by OCT2. |
|
| 参考文献 |
|
|
| 其他信息 |
Therapeutic Uses
Antineoplastic Caprelsa is a kinase inhibitor indicated for the treatment of symptomatic or progressive medullary thyroid cancer in patients with unresectable locally advanced or metastatic disease. /Incuded in US product label/ Because of the risk of QT prolongation, torsades de pointes, and sudden death, the US Food and Drug Administration (FDA) required and has approved a Risk Evaluation and Mitigation Strategy (REMS) for vandetanib. Under the terms of the REMS program, vandetanib is available only under a restricted distribution program (Caprelsa REMS Program). Prescribers and pharmacies must be certified with the Caprelsa REMS Program before they can prescribe or dispense vandetanib. To be certified, prescribers must review the educational materials, agree to comply with the REMS requirements, and enroll in the program. Pharmacies that dispense vandetanib must enroll in the program, train their pharmacy staff to verify that each prescription is written by a certified prescriber before dispensing the drug to the patient, and agree to comply with the REMS requirements. Vandetanib is used for the treatment of symptomatic or progressive medullary thyroid cancer in patients with unresectable locally advanced or metastatic disease; vandetanib is designated an orphan drug by the US Food and Drug Administration (FDA) for the treatment of this cancer. Drug Warnings /BOXED WARNING/ WARNING: QT PROLONGATION, TORSADES DE POINTES, AND SUDDEN DEATH. Caprelsa can prolong the QT interval. Torsades de pointes and sudden death have occurred in patients receiving Caprelsa. Do not use Caprelsa in patients with hypocalcemia, hypokalemia, hypomagnesemia, or long QT syndrome. Correct hypocalcemia, hypokalemia and/or hypomagnesemia prior to Caprelsa administration. Monitor electrolytes periodically. Avoid drugs known to prolong the QT interval. Only prescribers and pharmacies certified with the restricted distribution program are able to prescribe and dispense Caprelsa. Vandetanib prolongs the QT interval in a concentration-dependent manner. Torsades de pointes, ventricular tachycardia, and sudden death have been reported in patients receiving vandetanib. In the phase 3 clinical study, patients randomized to receive vandetanib (300 once daily) had a mean increase in the QT interval (corrected for heart rate using Fridericia's formula (QTcF)) of 35 msec (range: 33-36 msec) from baseline; this increase in QTcF remained above 30 msec for the duration of the study (up to 2 years). In addition, an increase in QTcF of more than 60 msec from baseline occurred in 36% of patients receiving vandetanib, and QTcF exceeded 450 msec or 500 msec in 69 or 7% of patients, respectively. Interstitial Lung Disease (ILD) or pneumonitis, including fatalities, has occurred in patients treated with Caprelsa. Consider a diagnosis of ILD in patients presenting with non-specific respiratory signs and symptoms. Interrupt Caprelsa for acute or worsening pulmonary symptoms. Discontinue Caprelsa if ILD is confirmed. Ischemic cerebrovascular events, sometimes fatal, have been reported with vandetanib. In the phase 3 clinical study, ischemic cerebrovascular events were observed more frequently with vandetanib compared with placebo (1.3 versus 0%); all ischemic cerebrovascular events reported in this study were grade 3. Vandetanib should be discontinued in patients who experience a severe ischemic cerebrovascular event. The safety of resumption of vandetanib therapy after resolution of an ischemic cerebrovascular event has not been studied. For more Drug Warnings (Complete) data for Vandetanib (20 total), please visit the HSDB record page. Pharmacodynamics Mean IC50 of approximately 2.1 μg/mL. |
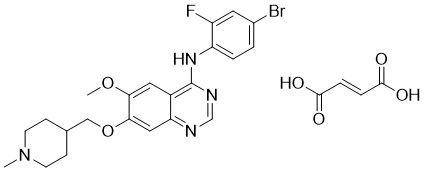
| 分子式 |
C26H28BRFN4O6
|
|---|---|
| 分子量 |
475.35396
|
| 精确质量 |
474.106
|
| 元素分析 |
C, 52.80; H, 4.77; Br, 13.51; F, 3.21; N, 9.47; O, 16.23
|
| CAS号 |
338992-00-0
|
| 相关CAS号 |
443913-73-3;338992-00-0 (fumarate); 338992-00-0; 338992-53-3; 524722-52-9
|
| PubChem CID |
3081361
|
| 外观&性状 |
Typically exists as solid at room temperature
|
| 密度 |
1.4±0.1 g/cm3
|
| 沸点 |
538.2±50.0 °C at 760 mmHg
|
| 闪点 |
279.3±30.1 °C
|
| 蒸汽压 |
0.0±1.4 mmHg at 25°C
|
| 折射率 |
1.629
|
| LogP |
5.51
|
| tPSA |
59.51
|
| 氢键供体(HBD)数目 |
1
|
| 氢键受体(HBA)数目 |
7
|
| 可旋转键数目(RBC) |
6
|
| 重原子数目 |
30
|
| 分子复杂度/Complexity |
539
|
| 定义原子立体中心数目 |
0
|
| InChi Key |
NEJYMTWEAISFSJ-WLHGVMLRSA-N
|
| InChi Code |
InChI=1S/C22H24BrFN4O2.C4H4O4/c1-28-7-5-14(6-8-28)12-30-21-11-19-16(10-20(21)29-2)22(26-13-25-19)27-18-4-3-15(23)9-17(18)245-3(6)1-2-4(7)8/h3-4,9-11,13-14H,5-8,12H2,1-2H3,(H,25,26,27)1-2H,(H,5,6)(H,7,8)/b2-1+
|
| 化学名 |
N-(4-Bromo-2-fluorophenyl)-6-methoxy-7-[(1-methylpiperidin-4-yl)methoxy]quinazolin-4-amine Fumarate
|
| 别名 |
Caprelsa; HSDB 8198; Zactima; ZD-6474; Vandetanib; 443913-73-3; Zactima; ZD6474; Caprelsa; N-(4-Bromo-2-fluorophenyl)-6-methoxy-7-((1-methylpiperidin-4-yl)methoxy)quinazolin-4-amine; ZD-6474; N-(4-bromo-2-fluorophenyl)-6-methoxy-7-[(1-methylpiperidin-4-yl)methoxy]quinazolin-4-amine; ZD 6474; ZD6474
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples
|
|---|---|
| 溶解度 (体内实验) |
注意: 如下所列的是一些常用的体内动物实验溶解配方,主要用于溶解难溶或不溶于水的产品(水溶度<1 mg/mL)。 建议您先取少量样品进行尝试,如该配方可行,再根据实验需求增加样品量。
注射用配方
注射用配方1: DMSO : Tween 80: Saline = 10 : 5 : 85 (如: 100 μL DMSO → 50 μL Tween 80 → 850 μL Saline)(IP/IV/IM/SC等) *生理盐水/Saline的制备:将0.9g氯化钠/NaCl溶解在100 mL ddH ₂ O中,得到澄清溶液。 注射用配方 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (如: 100 μL DMSO → 400 μL PEG300 → 50 μL Tween 80 → 450 μL Saline) 注射用配方 3: DMSO : Corn oil = 10 : 90 (如: 100 μL DMSO → 900 μL Corn oil) 示例: 以注射用配方 3 (DMSO : Corn oil = 10 : 90) 为例说明, 如果要配制 1 mL 2.5 mg/mL的工作液, 您可以取 100 μL 25 mg/mL 澄清的 DMSO 储备液,加到 900 μL Corn oil/玉米油中, 混合均匀。 View More
注射用配方 4: DMSO : 20% SBE-β-CD in Saline = 10 : 90 [如:100 μL DMSO → 900 μL (20% SBE-β-CD in Saline)] 口服配方
口服配方 1: 悬浮于0.5% CMC Na (羧甲基纤维素钠) 口服配方 2: 悬浮于0.5% Carboxymethyl cellulose (羧甲基纤维素) 示例: 以口服配方 1 (悬浮于 0.5% CMC Na)为例说明, 如果要配制 100 mL 2.5 mg/mL 的工作液, 您可以先取0.5g CMC Na并将其溶解于100mL ddH2O中,得到0.5%CMC-Na澄清溶液;然后将250 mg待测化合物加到100 mL前述 0.5%CMC Na溶液中,得到悬浮液。 View More
口服配方 3: 溶解于 PEG400 (聚乙二醇400) 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.1037 mL | 10.5186 mL | 21.0371 mL | |
| 5 mM | 0.4207 mL | 2.1037 mL | 4.2074 mL | |
| 10 mM | 0.2104 mL | 1.0519 mL | 2.1037 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
SAFIR02_Lung - Efficacy of Targeted Drugs Guided by Genomic Profiles in Metastatic NSCLC Patients
CTID: NCT02117167
Phase: Phase 2 Status: Completed
Date: 2024-01-10
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1


































