| 规格 | 价格 | |
|---|---|---|
| 500mg | ||
| 1g | ||
| Other Sizes |
| 靶点 |
HSV-1 (IC50 = 2.9 μg/mL)
|
|---|---|
| 体外研究 (In Vitro) |
根据 Michaelis-Menten 常数,Valaciclovir (VACV) 的最大速率分别为 23.34 nmol/mg 蛋白质/5 分钟和 1.64 mM,具有浓度依赖性和饱和吸收。大鼠、兔和 Caco-2 细胞以及 hPEPT1/CHO 细胞中的 Km 值非常相似,表明 hPEPT1 控制 VACV 的体外肠道转运特性 [5]。
|
| 体内研究 (In Vivo) |
一项重要的比较试验发现,对于治疗首次发作的生殖器疱疹,伐昔洛韦(1 g,每天两次)持续 10 天与阿昔洛韦(200 mg,每天 5 次)一样有效。两项试验发现,在为期五天的治疗周期中,伐昔洛韦(200 毫克,每日五次)与阿昔洛韦(200 毫克,每日五次)对于控制复发同样有效。伐昔洛韦每天 1 克的剂量与每天 2 克的效果相同。每天可以给予一剂伐昔洛韦[1]。口服伐昔洛韦 1,000 mg,每天 3 次,六天后,在稳态下评估血清和脑脊液阿昔洛韦浓度 [2]。 PE 和 AC 在 3T3 细胞中的 EC50 值分别为 0.02 和 0.01 ug/ml,但在 BHK 细胞中分别为 0.2 和 0.03 ug/ml。用FA和VA治疗感染的免疫抑制小鼠(每天两次,5.5天)以消除耳轻瘫、耳部病变(水泡等)和死亡。红斑比例也从 100% 降低至 24% 和 38%。到第六天,病毒已经从耳朵和脑干消失,但在接受 VA 治疗的小鼠中,当药物停止时,病毒又回来了 [3]。
|
| 酶活实验 |
通过使用斑块减少测定法测定HSV-1W菌株的体外50%抑制浓度(IC50),以验证其对阿昔洛韦的敏感性。HSV-1 W的IC50测定为2.9µg/ml[4]。
|
| 细胞实验 |
我们实验室先前使用原位灌注技术在大鼠和兔顶端刷状边界膜囊泡中进行的研究结果表明,缬环鸟苷(VACV)的肠道摄取似乎是由多种膜转运蛋白介导的。使用这些技术,在存在多种已知或未知转运蛋白的情况下,很难表征VACV与每个单独转运蛋白的转运动力学。本研究的目的是使用过表达人肠肽转运蛋白(hPEPT1)基因的中国仓鼠卵巢(CHO)细胞来表征VACV与人肠肽转运体的相互作用。用hPEPT1转染的CHO细胞中的VACV摄取显著高于仅用载体pcDNA3转染的细胞。VACV吸收的最佳pH确定为在pH 7.5时发生。在hPEPT1/CHO细胞中未观察到质子共转运,这与先前在组织和Caco-2细胞中观察到的结果一致。VACV摄取是浓度依赖性的,并且可饱和,米氏常数和最大速度分别为1.64+/-0.06mM和23.34+/-0.36nmol/mg蛋白质/5min。在hPEPT1/CHO细胞、大鼠和兔组织以及Caco-2细胞中获得了非常相似的Km值,表明hPEPT1在体外主导VACV的肠道运输特性。VACV的摄取被各种二肽和β-内酰胺类抗生素显著抑制,在pH 7.5时,Gly-Sar和头孢羟氨苄的Ki值分别为12.8+/-2.7和9.1+/-1.2mM。目前的结果表明,VACV是hPEPT1/CHO细胞中人类肠肽转运蛋白的底物,并且尽管转运是pH依赖性的,但质子协同转运并不明显。此外,结果表明hPEPT1/CHO细胞系统可用于研究药物与人肠肽转运蛋白hPEPT1的转运动力学;然而,将这些转运特性外推到体内情况需要进一步研究[5]。
|
| 动物实验 |
Acyclovir has been a frequently used antiviral agent in the clinical treatment of leukemia, acute encephalitis, malignant tumor and herpes simplex. The adverse effects of this drug have been widely described in clinical practice. In the present study, a case of a 35-year-old female patient diagnosed with herpes simplex, who developed acute renal injury following treatment with valacyclovir hydrochloride, is described. Kidney biopsy, light microscopy and laboratory examination were performed, and all findings revealed the signs of evident vacuolar degeneration of capillary endothelial and renal tubular epithelial cells, erythrocyte aggregation in partial renal tubule and microvilli exfoliation from epithelial cells. Renal interstitial edema was clearly identified. The clinical evidence observed from this female patient indicated that renal functions should be closely monitored during valacyclovir hydrochloride administration. A variety of effective measures, such as hydration, alkalizing urine, promoting the discharge of medication and the use of antagonists are recommended following the administration of antiviral agents[1].
|
| 药代性质 (ADME/PK) |
Absorption, Distribution and Excretion
After oral administration, valacyclovir hydrochloride is rapidly absorbed from the gastrointestinal (GI) tract and converted to acyclovir and L-valine. The absolute bioavailability of acyclovir after administration of valacyclovir was measured at 54.5% ± 9.1% after the administration of a 1 gram oral dose of valacyclovir and a 350 mg intravenous (IV) acyclovir dose to 12 healthy subjects. Acyclovir (a metabolite of valacyclovir) bioavailability from the administration of this drug is not affected by the administration with food. After oral administration of a single 1 gram dose of radiolabeled valacyclovir to 4 healthy subjects, 46% and 47% of administered radioactivity was measured in urine and feces, respectively, over 96 hours. Acyclovir accounted for 89% of the radioactivity excreted in the urine. Cerebrospinal fluid (CSF) penetration, determined by CSF/plasma AUC ratio, is approximately 25% for aciclovir and the metabolite _8-hydroxy-aciclovir_ (8-OH-ACV), and approximately 2.5% for the metabolite _9-(carboxymethoxy)methylguanine_. In a study of immunocompromised pediatric patients, the volume of distribution of a 15 ml/kg dose of valacyclovir was 1.34 ± 0.65 L/kg. Renal clearance of acyclovir following the administration of a single 1 gram dose of valacylcovir to 12 healthy 437 volunteers was approximately 255 ± 86 mL/min, which represents 42% of total acyclovir apparent plasma clearance. After oral administration, valacyclovir hydrochloride is rapidly absorbed from the gastrointestinal tract and nearly completely converted to acyclovir and L-valine by first-pass intestinal and/or hepatic metabolism. The absolute bioavailability of acyclovir after administration of Valtrex is 54.5% + or - 9.1% as determined following a 1-gram oral dose of Valtrex and a 350 mg intravenous acyclovir dose to 12 healthy volunteers. Acyclovir bioavailability from the administration of Valtrex is not altered by administration with food (30 minutes after an 873 Kcal breakfast, which included 51 grams of fat). The binding of valacyclovir to human plasma proteins ranges from 13.5% to 17.9%. The binding of acyclovir to human plasma proteins ranges from 9% to 33%. The pharmacokinetic disposition of acyclovir delivered by valacyclovir is consistent with previous experience from intravenous and oral acyclovir. Following the oral administration of a single 1 gram dose of radiolabeled valacyclovir to 4 healthy subjects, 46% and 47% of administered radioactivity was recovered in urine and feces, respectively, over 96 hours. Acyclovir accounted for 89% of the radioactivity excreted in the urine. Renal clearance of acyclovir following the administration of a single 1-gram dose of Valtrex to 12 healthy volunteers was approximately 255 + or - 86 mL/min which represents 42% of total acyclovir apparent plasma clearance. The mechanism of intestinal transport of valacyclovir, the L-valyl ester prodrug of acyclovir, was investigated in rats using an in situ intestinal perfusion technique. Results showed that the oral bioavailability of valacyclovir appears to be significantly influenced by the preabsorptive conversion of valacyclovir to the poorly absorbed acyclovir, by the involvement of multiple transporters in valacyclovir small-intestinal uptake, and by the low permeability of valacyclovir in the colon. Following oral administration of a 500 mg dose of VALTREX to 5 nursing mothers, peak acyclovir concentrations (Cmax) in breast milk ranged from 0.5 to 2.3 times (median 1.4) the corresponding maternal acyclovir serum concentrations. The acyclovir breast milk AUC ranged from 1.4 to 2.6 times (median 2.2) maternal serum AUC. A 500 mg maternal dosage of VALTREX twice daily would provide a nursing infant with an oral acyclovir dosage of approximately 0.6 mg/kg/day. This would result in less than 2% of the exposure obtained after administration of a standard neonatal dose of 30 mg/kg/day of intravenous acyclovir to the nursing infant. Unchanged valacyclovir was not detected in maternal serum, breast milk, or infant urine. Metabolism / Metabolites Valacyclovir is converted to acyclovir and L-valine via first-pass intestinal and/or hepatic metabolism. Acyclovir is also transformed, to a small extent, to inactive metabolites by _aldehyde oxidase_ in addition to _alcohol dehydrogenase_ and _aldehyde dehydrogenase_. Neither valacyclovir nor acyclovir is metabolized by cytochrome P450 enzymes. ... Aciclovir's main metabolite /is/ 9-carboxymethoxymethylguanine. ... Valacyclovir is converted to acyclovir and L-valine by first-pass intestinal and/or hepatic metabolism. Acyclovir is converted to a small extent to inactive metabolites by aldehyde oxidase and by alcohol and aldehyde dehydrogenase. Neither valacyclovir nor acyclovir is metabolized by cytochrome P450 enzymes. Plasma concentrations of unconverted valacyclovir are low and transient, generally becoming non-quantifiable by 3 hours after administration. Peak plasma valacyclovir concentrations are generally less than 0.5 ug/mL at all doses. After single-dose administration of 1 gram of Valtrex, average plasma valacyclovir concentrations observed were 0.5, 0.4, and 0.8 ug/mL in patients with hepatic dysfunction, renal insufficiency, and in healthy volunteers who received concomitant cimetidine and probenecid, respectively. Biological Half-Life The plasma elimination half-life of acyclovir typically averaged 2.5 to 3.3 hours in several studies of valacyclovir in volunteers with normal renal function. The plasma elimination half-life of acyclovir typically averaged 2.5 to 3.3 hours in all studies of Valtrex in volunteers with normal renal function. |
| 毒性/毒理 (Toxicokinetics/TK) |
Hepatotoxicity
Oral therapy with valacyclovir is associated with a low rate of mild-to-moderate serum aminotransferase elevations, but these abnormalities are usually asymptomatic and self-limited even with continuation of therapy. Complicating the attribution of liver test abnormalities to valacyclovir therapy is the fact that enzyme elevations are not uncommon during the course of varicella-zoster infection (both chickenpox and shingles) and can progress to clinically apparent hepatitis and even acute liver failure. Clinically apparent liver disease due to valacyclovir itself is rare, but isolated reports have been published. The time to onset was short (1 to 2 weeks) and the course mild, with few symptoms and rapid resolution (Case 1). The pattern of liver injury described was mixed hepatocellular-cholestatic. Immunoallergic features and autoantibodies were absent. Likelihood score: D (possible rare cause of clinically apparent liver injury). Effects During Pregnancy and Lactation ◉ Summary of Use during Lactation The dosage of acyclovir in milk after valacyclovir is less than 1% of a typical infant dosage and would not be expected to cause any adverse effects in breastfed infants. No special precautions are required when using valacyclovir during breastfeeding. In one study, administration of valacyclovir to mothers with concurrent herpes simplex type 2 and HIV infections reduced breastmilk shedding of the HIV virus in breastmilk at 6 and 14 weeks postpartum, but not later.[1] In another study in HIV-positive mothers, valacyclovir did not reduced breastmilk shedding of cytomegalovirus (CMV) or infant CMV acquisition.[2] ◉ Effects in Breastfed Infants In a study of pregnant women with concurrent HIV and Herpes simplex infections, mothers received zidovudine 300 mg daily from week of pregnancy until 12 months postpartum and nevirapine at delivery. Half of the women (n = 74) also received valacyclovir 500 mg orally twice daily from 34 weeks gestation until 12 months postpartum. At 6 weeks postpartum, all infants who received acyclovir in breastmilk had normal serum creatinine (<0.83 mg/dL). Their median serum creatinine and alanine aminotransferase (ALT) values, and growth were no different from those of unexposed infants, with the exception of one infant with an ALT level of 70.1 units/L. Infants whose mothers received valacyclovir generally had adverse effects that were similar to the placebo group, except that treated infants had a lower risk of eczema and oral thrush than infants in the placebo arm.[1][4] ◉ Effects on Lactation and Breastmilk Relevant published information was not found as of the revision date. Protein Binding The binding of valacyclovir to human plasma proteins is low and ranges from 13.5% to 17.9%. Interactions Concomitant use of valacyclovir and probenecid may increase peak plasma concentrations and AUC of acyclovir. This pharmacokinetic interaction is not considered clinically important in patients with normal renal function and no dosage adjustments are necessary in these patients. Concomitant use of valacyclovir and cimetidine may increase peak plasma concentrations and AUC of acyclovir. This pharmacokinetic interaction is not considered clinically important in patients with normal renal function; no dosage adjustments are necessary in these patients. Mycophenolate mofetil (MMF) is a drug which decreases the frequency of renal transplantation rejection. However, cytomegalovirus infections are a common feature of this treatment leading the physicians to prescribe antiviral prophylactic drugs like valacyclovir. During this association, neutropenia occur and the cause of this adverse effect is difficult to define. This report presents a case of neutropenia in a woman treated with MMF and valacyclovir. As the duration of the valacyclovir treatment exactly corresponds to the neutropenia duration, and the mycophenolate trough levels increased with the neutrophil count, the responsibility of this neutropenia was ascribed to valacyclovir. However, an examination of the literature for cases of neutropenia led to the suspicion of an interaction between MMF and valacyclovir. Mycophenolate may increase intracellular concentrations of valacyclovir up to hematotoxic levels. This mechanism may explain the interaction and further research is needed to confirm this interaction. |
| 参考文献 |
|
| 其他信息 |
Therapeutic Uses
Antiviral Agents Oral valacyclovir is used in the treatment of initial episodes of genital herpes simplex virus (HSV-2) infection in immunocompetent adults and adolescents. Because many patients with first episodes of genital herpes present with mild clinical symptoms but later develop severe or prolonged symptoms, the US Centers for Disease Control and Prevention (CDC) states that most patients with initial genital herpes should receive antiviral therapy. /Included in US product labeling/ Oral valacyclovir is used in the treatment of recurrent episodes of genital herpes in immunocompetent adults and adolescents. Antiviral therapy for recurrent genital herpes can be given episodically to ameliorate or shorten the duration of lesions or can be given continuously as suppressive therapy to reduce the frequency of recurrences. /Included in US product labeling/ Valacyclovir is used for the episodic treatment of herpes labialis (perioral herpes, cold sores, fever blisters) in adults and adolescents. /Included in US product labeling/ For more Therapeutic Uses (Complete) data for Valacyclovir (9 total), please visit the HSDB record page. Drug Warnings Thrombotic Thrombocytopenic Purpura/Hemolytic Uremic Syndrome (TTP/HUS), in some cases resulting in death, has occurred in patients with advanced HIV-1 disease and also in allogeneic bone marrow transplant and renal transplant recipients participating in clinical trials of Valtrex at doses of 8 grams per day. Treatment with Valtrex should be stopped immediately if clinical signs, symptoms, and laboratory abnormalities consistent with TTP/HUS occur. There are no adequate and well-controlled studies of Valtrex or acyclovir in pregnant women. Based on prospective pregnancy registry data on 749 pregnancies, the overall rate of birth defects in infants exposed to acyclovir in-utero appears similar to the rate for infants in the general population. Valtrex should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus. Cases of acute renal failure have been reported in: 1. Elderly patients with or without reduced renal function. Caution should be exercised when administering Valtrex to geriatric patients, and dosage reduction is recommended for those with impaired renal function. 2. Patients with underlying renal disease who received higher than recommended doses of Valtrex for their level of renal function. Dosage reduction is recommended when administering Valtrex to patients with renal impairment. 3. Patients receiving other nephrotoxic drugs. Caution should be exercised when administering Valtrex to patients receiving potentially nephrotoxic drugs. 4. Patients without adequate hydration. Precipitation of acyclovir in renal tubules may occur when the solubility (2.5 mg/mL) is exceeded in the intratubular fluid. Adequate hydration should be maintained for all patients. The most common adverse reactions reported in at least 1 indication by greater than 10% of adult patients treated with Valtrex and observed more frequently with Valtrex compared to placebo are headache, nausea, and abdominal pain. The only adverse reaction reported in greater than 10% of pediatric patients aged less than 18 years was headache. For more Drug Warnings (Complete) data for Valacyclovir (8 total), please visit the HSDB record page. Pharmacodynamics **Antiviral effects** Valacyclovir shows varying levels of inhibition towards herpes simplex virus types 1 (HSV-1), 2 (HSV-2), Varicella Zoster Virus (VZV), Epstein-Barr virus (EBV), and cytomegalovirus (CMV). The quantitative relationship between the cell culture susceptibility of herpesviruses to antivirals and the clinical response of humans to the same antiviral therapy has not yet been elucidated. Sensitivity testing results, described by the concentration of drug needed to inhibit the growth of the virus by 50% in cell culture (EC50), vary widely depending on various factors. **Clinical study results** For the various conditions below, clinical study results are summarized as follows: _Cold sores_ Immunocompetent volunteers with cold sores were observed following the administration of a 1-day regimen (2 grams of valacyclovir twice a day for 1 day followed by one day of placebo) or a 2-day regimen (2 grams of valacyclovir twice daily for two days). The average duration of cold sore episodes was approximately 1 day shorter in treated subjects when compared to subjects treated with placebo. A 2-day drug administration regimen of valacyclovir did not provide superior benefit over the 1-day regimen. There was no clinically significant difference observed between subjects receiving valacyclovir or placebo in the prevention of progression of cold sore lesions after the papular stage, indicating that timing of valacyclovir administration is an important consideration. _Initial genital herpes episodes_ 643 immunocompetent adults with first-episode genital herpes who presented within 72 hours of symptom onset were randomized in a double-blind trial to receive 10 days of valacyclovir 1 gram twice daily (n = 323) or oral acyclovir 200 mg 5 times a day (n = 320). In both groups, the median time to healing of herpetic lesions was measured to be 9 days, and the median time to cessation of pain was found to be 5 days, with the median time to cessation of viral shedding was approximately 3 days. _Recurrent genital herpes episodes_ The results of 3 separate studies of patients taking 3 to 5-day regimens of valacyclovir showed an average of 4 days to lesion healing, 2-3 days to resolution of pain associated with the lesions, with an average of 2 days until the cessation of viral shedding. These findings showed valacyclovir administration to show superior beneficial effects when compared to the findings associated with placebo administration. **A note on resistance** The resistance of Herpes Simplex Virus and Varicella Zoster Virus to acyclovir can result from qualitative and quantitative changes in the viral TK and/or DNA polymerase. Clinical isolates of VZV with decreased susceptibility to acyclovir have been isolated from patients diagnosed with AIDS. A total of 522 TK-deficient mutants of VZV have been identified in these cases. |
| 分子式 |
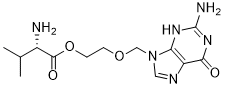
C13H20N6O4
|
|---|---|
| 分子量 |
324.341
|
| 精确质量 |
324.154
|
| 元素分析 |
C, 48.14; H, 6.22; N, 25.91; O, 19.73
|
| CAS号 |
124832-26-4
|
| 相关CAS号 |
Valacyclovir hydrochloride;124832-27-5;Valacyclovir hydrochloride hydrate;1218948-84-5
|
| PubChem CID |
135398742
|
| 外观&性状 |
Typically exists as solid at room temperature
|
| 密度 |
1.5±0.1 g/cm3
|
| 熔点 |
170-172
|
| 闪点 |
309.7ºC
|
| 折射率 |
1.673
|
| LogP |
-0.88
|
| tPSA |
151.14
|
| 氢键供体(HBD)数目 |
3
|
| 氢键受体(HBA)数目 |
7
|
| 可旋转键数目(RBC) |
8
|
| 重原子数目 |
23
|
| 分子复杂度/Complexity |
483
|
| 定义原子立体中心数目 |
1
|
| SMILES |
N[C@@H](C(C)C)C(OCCOCN1C=NC2=C1N=C(N)NC2=O)=O
|
| InChi Key |
HDOVUKNUBWVHOX-QMMMGPOBSA-N
|
| InChi Code |
InChI=1S/C13H20N6O4/c1-7(2)8(14)12(21)23-4-3-22-6-19-5-16-9-10(19)17-13(15)18-11(9)20/h5,7-8H,3-4,6,14H2,1-2H3,(H3,15,17,18,20)/t8-/m0/s1
|
| 化学名 |
2-((2-amino-6-oxo-1,6-dihydro-9H-purin-9-yl)methoxy)ethyl L-valinate
|
| 别名 |
BW-256U87; BW-256; BW256256U87 hydrochloride; BW 256 Val-ACV; Valtrex; Zelitrex; Valacyclovir HCl; Valacyclovir hydrochloride; ValACV; Zelitrex; Valcivir; Valcyclovir; Val-ACV;
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples
|
|---|---|
| 溶解度 (体内实验) |
注意: 如下所列的是一些常用的体内动物实验溶解配方,主要用于溶解难溶或不溶于水的产品(水溶度<1 mg/mL)。 建议您先取少量样品进行尝试,如该配方可行,再根据实验需求增加样品量。
注射用配方
注射用配方1: DMSO : Tween 80: Saline = 10 : 5 : 85 (如: 100 μL DMSO → 50 μL Tween 80 → 850 μL Saline)(IP/IV/IM/SC等) *生理盐水/Saline的制备:将0.9g氯化钠/NaCl溶解在100 mL ddH ₂ O中,得到澄清溶液。 注射用配方 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (如: 100 μL DMSO → 400 μL PEG300 → 50 μL Tween 80 → 450 μL Saline) 注射用配方 3: DMSO : Corn oil = 10 : 90 (如: 100 μL DMSO → 900 μL Corn oil) 示例: 以注射用配方 3 (DMSO : Corn oil = 10 : 90) 为例说明, 如果要配制 1 mL 2.5 mg/mL的工作液, 您可以取 100 μL 25 mg/mL 澄清的 DMSO 储备液,加到 900 μL Corn oil/玉米油中, 混合均匀。 View More
注射用配方 4: DMSO : 20% SBE-β-CD in Saline = 10 : 90 [如:100 μL DMSO → 900 μL (20% SBE-β-CD in Saline)] 口服配方
口服配方 1: 悬浮于0.5% CMC Na (羧甲基纤维素钠) 口服配方 2: 悬浮于0.5% Carboxymethyl cellulose (羧甲基纤维素) 示例: 以口服配方 1 (悬浮于 0.5% CMC Na)为例说明, 如果要配制 100 mL 2.5 mg/mL 的工作液, 您可以先取0.5g CMC Na并将其溶解于100mL ddH2O中,得到0.5%CMC-Na澄清溶液;然后将250 mg待测化合物加到100 mL前述 0.5%CMC Na溶液中,得到悬浮液。 View More
口服配方 3: 溶解于 PEG400 (聚乙二醇400) 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 3.0832 mL | 15.4159 mL | 30.8318 mL | |
| 5 mM | 0.6166 mL | 3.0832 mL | 6.1664 mL | |
| 10 mM | 0.3083 mL | 1.5416 mL | 3.0832 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
Prenatal Treatment of Congenital Cytomegalovirus Infection with Letermovir Versus Valaciclovir
CTID: NCT05446571
Phase: Phase 3 Status: Recruiting
Date: 2024-09-04
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1


































