| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 5mg |
|
||
| 10mg |
|
||
| Other Sizes |
|
| 体外研究 (In Vitro) |
曲美曲沙(0.1 μM,18 小时)完全抑制由弓形虫引起的小鼠巨噬细胞肿胀[3]。弓形虫细胞膜可渗透三甲曲酯 (1 μM),可快速达到细胞内高浓度(108 pmol/107 个细胞)。在 SNU-C4 和 NCI-H630 细胞系中,曲美曲沙(0.1 mM;24 小时)可降低细胞生长 50-60% [5]。十分钟内,添加 C4 中的曲美曲沙(1 和 10 mM;24 小时)[3]。
|
|---|---|
| 体内研究 (In Vivo) |
Trimetrexate(180 mg/kg 或 30 mg/kg;口服或腹腔注射;每日)可延长弓形虫感染的中位生存期,并显示出抗弓形虫活性 [3]。曲美曲沙(0-30 mg/kg;静脉注射;每天一次,持续 5 天)表现出慢性毒性[4]。
|
| 细胞实验 |
细胞增殖测定[5]
细胞类型: SNU-C4 和 NCI-H630 测试浓度: 0.1 mM 孵育时间: 24小时 实验结果:两种细胞系均抑制细胞生长50-60%。 细胞增殖测定[5] 细胞类型: C4 细胞 测试浓度: 1 和 10 mM 孵育持续时间:24 小时 实验结果:在 1 和 10 mM 浓度下分别产生 42% 和 50% 的致死率。 |
| 动物实验 |
Animal/Disease Models: Female balb/c (Bagg ALBino) mouse infected with Toxoplasma gondii, weighing about 20 g [3]
Doses: 180 mg/kg or 30 mg/kg Dosing: 180 mg/kg orally daily in drinking water or intraperitoneal (ip) injection 30 times daily mg/kg Experimental Results: Extended median survival of infected mice to 10 days (oral) or 19 days (ip). Animal/Disease Models: Charles River Wistar Crl(WI)BR rats, weighing approximately 150 to 200 g[4] Doses: 0, 1, 10 or 30 mg/kg Route of Administration: intravenous (iv) (iv)injection, one time/day for 5 days, then 23- Experimental Results: Showing chronic toxicity, testicular changes that persisted over the course of multiple dosing cycles were irreversible within 21 days but required an additional 56 days for essentially complete recovery. |
| 药代性质 (ADME/PK) |
Absorption, Distribution and Excretion
Ten to 30% of the administered dose is excreted unchanged in the urine. 20 ± 8 L/m2 36.9 ± 6 L/m2 [cancer patients] 38 +/- 15 mL/min/m2 [patients with acquired immunodeficiency syndrome (AIDS) who had Pneumocystis carinii pneumonia (4 patients) or toxoplasmosis (2 patients). Trimetrexate was administered intravenously as a bolus injection at a dose of 30 mg/m2/day along with leucovorin 20 mg/m2 every 6 hours for 21 days] 53 +/- 41 mL/min/m2 [Cancer patients with advanced solid tumors using various dosage regimensreceiving a single-dose administration of 10 to 130 mg/m2] 30 +/- 8 mL/min/m2 [Cancer patients with advanced solid tumors using various dosage regimensafter a five-day infusion] Clinical pharmacokinetic studies in cancer patients show that trimetrexate plasma concentration-time curves are biphasic or triphasic in form. The terminal elimination half-life averages 13.6 hr. Mean total plasma clearance and volume of distribution as steady-state values were 27.9 ml/min/sq m and 21.1 l/sq m, respectively. Cerebrospinal fluid concentration was 3.4% of that in plasma, which shows that trimetrexate does not cross the blood-brain barrier well. Trimetrexate is 86 to 94% bound to plasma proteins. Mean oral bioavailability of the parenteral solution (glucuronate) in AIDS patients was 42%, with a mean maximum plasma concentration of 1182 ng/ml (3.2 umole/l) achieved 1.8 hr postdose. /Trimetrexate glucuromate/ Metabolism / Metabolites Hepatic. Preclinical data strongly suggest that the major metabolic pathway is oxidative O-demethylation, followed by conjugation to either glucuronide or the sulfate. Trimetrexate is highly metabolized by the liver. At least 2 metabolites are excreted in urine. One metabolite has been identified a 4'-O-glucuronide conjugate of trimetrexate, which is formed as a result of oxidative O-dimethylation at the 4'-position and subsequent conjugation with glucuronic acid. About 15% of a dose is excreted in urine as unchanged trimetrexate, while another 20% apparently excreted as metabolites. Hepatic. Preclinical data strongly suggest that the major metabolic pathway is oxidative O-demethylation, followed by conjugation to either glucuronide or the sulfate. Route of Elimination: Ten to 30% of the administered dose is excreted unchanged in the urine. Half Life: 11 to 20 hours Biological Half-Life 11 to 20 hours The terminal elimination half-life averages 13.6 hours. |
| 毒性/毒理 (Toxicokinetics/TK) |
Toxicity Summary
In vitro studies have shown that trimetrexate is a competitive inhibitor of dihydrofolate reductase (DHFR) from bacterial, protozoan, and mammalian sources. DHFR catalyzes the reduction of intracellular dihydrofolate to the active coenzyme tetrahydrofolate. Inhibition of DHFR results in the depletion of this coenzyme, leading directly to interference with thymidylate biosynthesis, as well as inhibition of folate-dependent formyltransferases, and indirectly to inhibition of p.r.n. biosynthesis. The end result is disruption of DNA, RNA, and protein synthesis, with consequent cell death. Hepatotoxicity When given without leucovorin protection, trimetrexate therapy is associated with a moderate rate of serum enzyme elevations, serum ALT or AST elevations above 5 times ULN in up to 20% of patients. When given with leucovorin, however, trimetrexate has fewer side effects although serum enzyme elevations can still occur. In clinical trials in patients with HIV infection and Pneumocystis jirovecii pneumonia, ALT elevations above 5 times ULN occurred in 1% to 8% of patients, but were usually no more frequent than with standard therapy using trimethoprim with sulfamethoxazole. The elevations were typically transient, without accompanying symptoms or jaundice and resolved or improved despite continuation of therapy. No instances of clinically apparent acute liver injury attributed to trimetrexate have been reported in the literature. In addition, trimetrexate has not been linked to sinusoidal obstruction syndrome or to reactivation of hepatitis B. Nevertheless, trimetrexate probably has hepatotoxic potential, but because it has limited use, is given for short periods of time and is administered with leucovorin, it has not been convincingly linked to cases of clinically apparent liver injury with jaundice. Likelihood score: E* (unproven but suspected cause of liver injury). Protein Binding 95% (over the concentration range of 18.75 to 1000 ng/mL) Toxicity Data LD50: 62 mg/kg (Intravenous, Mouse) (A308) |
| 参考文献 |
[1]. Hopper AT, et al. Discovery of Selective Toxoplasma gondii Dihydrofolate Reductase Inhibitors for the Treatment of Toxoplasmosis. J Med Chem. 2019 Feb 14;62(3):1562-1576.
[2]. Fulton, B., et al. Trimetrexate. Drugs 49, 563–576 (1995). [3]. Allegra CJ, et al. Potent in vitro and in vivo antitoxoplasma activity of the lipid-soluble antifolate trimetrexate. J Clin Invest. 1987 Feb;79(2):478-82. [4]. Dethloff LA, et al. Chronic toxicity of the anticancer agent trimetrexate in rats. Fundam Appl Toxicol. 1992 Jul;19(1):6-14. [5]. Grem JL, Voeller DM, Geoffroy F, Horak E, Johnston PG, Allegra CJ. Determinants of trimetrexate lethality in human colon cancer cells. Br J Cancer. 1994 Dec;70(6):1075-84. |
| 其他信息 |
Trimetrexate is a member of quinazolines. It has a role as an antifungal drug.
A nonclassical folic acid inhibitor through its inhibition of the enzyme dihydrofolate reductase. It is being tested for efficacy as an antineoplastic agent and as an antiparasitic agent against pneumocystis pneumonia in AIDS patients. Myelosuppression is its dose-limiting toxic effect. Trimetrexate is a parenterally administered folate antagonist that is used as a second line therapy for severe Pneumocystis jirovecii (previously carinii) pneumonia. Trimetrexate therapy has been associated with transient, mild serum enzyme elevations during therapy, but has not been convincingly linked to instances of acute, clinically apparent liver injury. Trimetrexate is a methotrexate derivative with potential antineoplastic activity. Trimetrexate inhibits the enzyme dihydrofolate reductase, thereby preventing the synthesis of purine nucleotides and thymidylate, with subsequent inhibition of DNA and RNA synthesis. Trimetrexate also exhibits antiviral activity. (NCI04) A nonclassical folic acid inhibitor through its inhibition of the enzyme dihydrofolate reductase. It is being tested for efficacy as an antineoplastic agent and as an antiparasitic agent against pneumocystis pneumonia in AIDS patients. Myelosuppression is its dose-limiting toxic effect. [PubChem] A nonclassical folic acid inhibitor through its inhibition of the enzyme dihydrofolate reductase. It is being tested for efficacy as an antineoplastic agent and as an antiparasitic agent against PNEUMOCYSTIS PNEUMONIA in AIDS patients. Myelosuppression is its dose-limiting toxic effect. Drug Indication For use, with concurrent leucovorin administration (leucovorin protection), as an alternative therapy for the treatment of moderate-to-severe Pneumocystis carinii pneumonia (PCP) in immunocompromised patients, including patients with the acquired immunodeficiency syndrome (AIDS). Also used to treat several types of cancer including colon cancer. FDA Label Mechanism of Action In vitro studies have shown that trimetrexate is a competitive inhibitor of dihydrofolate reductase (DHFR) from bacterial, protozoan, and mammalian sources. DHFR catalyzes the reduction of intracellular dihydrofolate to the active coenzyme tetrahydrofolate. Inhibition of DHFR results in the depletion of this coenzyme, leading directly to interference with thymidylate biosynthesis, as well as inhibition of folate-dependent formyltransferases, and indirectly to inhibition of p.r.n. biosynthesis. The end result is disruption of DNA, RNA, and protein synthesis, with consequent cell death. Trimetrexate binds to dihydrofolate reductase and prevents the conversion of dihydrofolate to biologically active tetrahydrofolate. It inhibits nucleic acid synthesis as a result of antithymidylate and antipurine effects. Therapeutic Uses Antifungal Agents; Antimetabolites; Antimetabolites, Antineoplastic; Folic Acid Antagonists Antineoplastic Trimetrexate glucuronate is an investigational drug that is available for treatment use ... in a hospital setting in qualifying patients with Pneumocystis carinii pneumonia and who have exhibited serious ... intolerance to both co-trimoxazole and pentamidine. /Trimetrexate glucuronate/ Drug Warnings Even though trimetrexate does not compete with the folate transport system for entry into cells, utilization of folates is reduced due to inhibition of dihydrofolate reductase. When trimetrexate is administered in high doses for the treatment of pneumocystis carinii pneumonia, leucovorin must be given concurrently with trimetrexate to reduce toxic effects on human tissues. It has been postulated that the absence of a folate transport system in pneumocystis carinii provides the opportunity for differential rescue of host tissue affecting antiprotozoal action. Patients receiving trimetrexate should have frequent laboratory monitoring of hepatic and hematologic prameters, including serum alanine aminotransferase, serum aspartate aminotransferase, bilirubin, alkaline phosphatase, platelet count, and total and differential leukocyte counts. Although uncommon in AIDS patients receiving trimetrexate for pneumocystis carinii pneumonia, slight elevations of blood urea nitrogen and/or serum creatinine have been reported in patients receiving the drug for various cancers. Isolated perfused rat liver has been used to study potential drug interactions with trimetrexate metabolism in vitro. Cimetidine, which inhibits oxidative drug metabolizing enzymes, decreased the clearance of trimetrexate to approximately one half of control values. Whether this occurs to a significant extent in vivo is unknown. Pharmacodynamics Trimetrexate, a non-classical folate antagonist, is a synthetic inhibitor of the enzyme dihydrofolate reductase (DHFR). During DNA synthesis and cellular reproduction, folic acid is reduced to tetrahydrofolic acid by the enzyme folic acid reductase. By interfering with the reduction of folic acid, trimetrexate interferes with tissue cell reproduction. Generally, the most sensitive cells to the antimetabolite effect of trimetrexate are those cells which are most actively proliferating such as malignant cells, dermal epithelium, buccal and intestinal mucosa, bone marrow, fetal cells, and cells of the urinary bladder. Because the proliferation of cells in malignant tissues is greater than in most normal tissues, trimetrexate may impair the growth of the malignant tissues without causing irreversible damage to normal tissues. Due to very serious and potentially life-threatening side-effects of this drug, leucovorin must be co-administered for at least 72 hours after the last dose. |
| 分子式 |
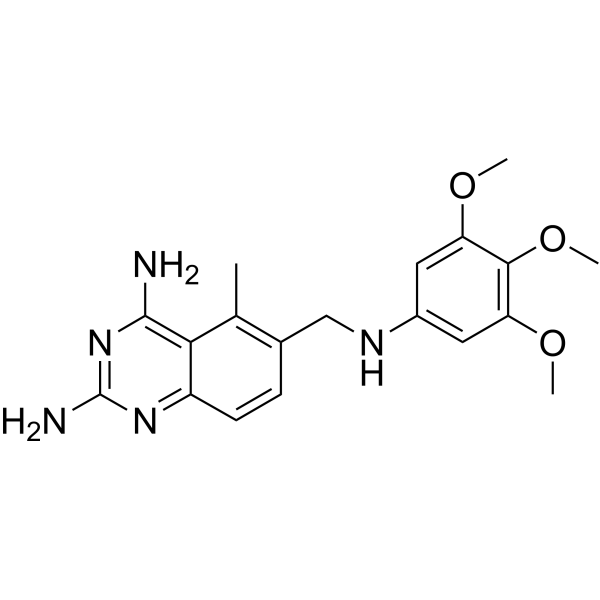
C19H23N5O3
|
|---|---|
| 分子量 |
369.41762
|
| 精确质量 |
369.18
|
| CAS号 |
52128-35-5
|
| 相关CAS号 |
Trimetrexate glucuronate;82952-64-5;Trimetrexate trihydrochloride;1658520-97-8;Trimetrexate isethionate;82935-04-4
|
| PubChem CID |
5583
|
| 外观&性状 |
Typically exists as solid at room temperature
|
| 密度 |
1.305g/cm3
|
| 沸点 |
647ºC at 760mmHg
|
| 熔点 |
215-217 °C
215 - 217 °C |
| 闪点 |
345.1ºC
|
| LogP |
3.975
|
| tPSA |
117.54
|
| 氢键供体(HBD)数目 |
3
|
| 氢键受体(HBA)数目 |
8
|
| 可旋转键数目(RBC) |
6
|
| 重原子数目 |
27
|
| 分子复杂度/Complexity |
457
|
| 定义原子立体中心数目 |
0
|
| SMILES |
NC1=C(C(C)=C2CNC3=CC(OC)=C(OC)C(OC)=C3)C(C=C2)=NC(N)=N1
|
| InChi Key |
NOYPYLRCIDNJJB-UHFFFAOYSA-N
|
| InChi Code |
InChI=1S/C19H23N5O3/c1-10-11(5-6-13-16(10)18(20)24-19(21)23-13)9-22-12-7-14(25-2)17(27-4)15(8-12)26-3/h5-8,22H,9H2,1-4H3,(H4,20,21,23,24)
|
| 化学名 |
5-methyl-6-[(3,4,5-trimethoxyanilino)methyl]quinazoline-2,4-diamine
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
DMSO : ≥ 61.5 mg/mL (~166.48 mM)
|
|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 2.5 mg/mL (6.77 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 25.0 mg/mL澄清DMSO储备液加入到400 μL PEG300中,混匀;然后向上述溶液中加入50 μL Tween-80,混匀;加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: ≥ 2.5 mg/mL (6.77 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 25.0 mg/mL 澄清 DMSO 储备液加入到 900 μL 玉米油中并混合均匀。 View More
配方 3 中的溶解度: 40 mg/mL (108.28 mM) in 50% PEG300 50% Saline (这些助溶剂从左到右依次添加,逐一添加), 悬浊液; 超声助溶。 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.7069 mL | 13.5347 mL | 27.0695 mL | |
| 5 mM | 0.5414 mL | 2.7069 mL | 5.4139 mL | |
| 10 mM | 0.2707 mL | 1.3535 mL | 2.7069 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1


































