| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| 500mg |
|
||
| 1g |
|
||
| 2g |
|
||
| Other Sizes |
|

| 靶点 |
HSV-2; HSV-1; Nucleoside Antimetabolite/Analog; Thymidylate Synthase
|
||
|---|---|---|---|
| 体外研究 (In Vitro) |
体外活性:曲氟尿苷对裸鼠移植的小鼠骨髓细胞和人结直肠癌细胞的增殖有剂量依赖性抑制作用。曲氟尿苷以浓度依赖性方式抑制骨髓细胞集落形成。由于 TK1 和 DUT 的底物特异性不同,三氟尿苷 (FTD) 和 2-脱氧-5-氟尿苷 (FdUrd) 以不同的效率掺入 DNA,导致大量 FTD 掺入 DNA。与 FdUrd 处理的细胞相比,FTD 处理的细胞显示出不同的核形态。曲氟尿苷对裸鼠移植的小鼠骨髓细胞和人结直肠癌细胞的增殖具有剂量依赖性抑制作用。
|
||
| 体内研究 (In Vivo) |
与未经治疗的受感染兔子的眼睛相比,曲氟尿苷中 HSV-1 阳性的拭子数量显着减少。 Trifluridine 具有更高的抗肿瘤活性,并且比在小鼠中持续输注 Trifluridine 更有效地掺入 DNA。与 5-FU 衍生物治疗相比,曲氟尿苷以不依赖 TPI 的方式逐渐在肿瘤细胞 DNA 中积累,并显着延迟肿瘤生长并延长小鼠的生存期。在新西兰兔眼模型中,曲氟尿苷可显着降低病毒滴度、治疗期间 HSV-1 阳性眼/总眼数、角膜炎评分较低、患有角膜炎眼数/总眼数以及角膜炎消退时间更短。
|
||
| 动物实验 |
|
||
| 药代性质 (ADME/PK) |
Absorption, Distribution and Excretion
After oral administration of LONSURF with [14C]-trifluridine, at least 57% of the administered trifluridine was absorbed. Following a single dose of LONSURF (35 mg/m2) in patients with advanced solid tumors, the mean times to peak plasma concentrations (Tmax) of trifluridine was around 2 hours. Trifluridine area under the concentration-time curve from time 0 to the last measurable concentration (AUC0-last) was approximately 3-fold higher and maximum concentration (Cmax) was approximately 2-fold higher after multiple dose administration (twice daily for 5 days a week with 2 days rest for 2 weeks followed by a 14-day rest, repeated every 4 weeks) than after single-dose administration. Following a single oral administration of LONSURF at 35 mg/m2 in patients with cancer, the mean time to peak plasma concentration (Tmax) of trifluridine was around 2 hours. For the ophthalmic formulation, systemic absorption appears to be negligible. A standardized high-fat, high-calorie meal decreased trifluridine Cmax by approximately 40% but did not change trifluridine AUC compared to those in a fasting state in patients with cancer following administration of a single dose of LONSURF 35 mg/m2. In a dose finding study (15 to 35 mg/m2 twice daily), the AUC from time 0 to 10 hours (AUC0-10) of trifluridine tended to increase more than expected based on the increase in dose. After single oral administration of LONSURF (60 mg) with [14C]-trifluridine, the total cumulative excretion of radioactivity was 60% of the administered dose. The majority of recovered radioactivity was eliminated into urine (55% of the dose) as FTY and trifluridine glucuronide isomers within 24 hours and the excretion into feces and expired air was <3% for both. The unchanged trifluridine was <3% of administered dose recovered in the urine and feces. Following a single dose of LONSURF (35 mg/m2) in patients with advanced solid tumours, the apparent volume of distribution (Vd/F) for trifluridine was 21 L. Following a single dose of LONSURF (35 mg/m2) in patients with advanced solid tumours, the oral clearance (CL/F) for trifluridine was 10.5 L/hr. Following topical application of trifluridine to the eye, the drug penetrates the cornea and can be detected in the aqueous humor. Systemic absorption following ocular application of trifluridine appears to be negligible. In one study in healthy individuals, topical application of trifluridine 1% ophthalmic solution to the eyes 7 times daily for 14 consecutive days did not result in detectable serum concentrations of trifluridine or 5-carboxy-2'-deoxyuridine. During in vitro studies using excised rabbit corneas, the major metabolite of trifluridine, 5-carboxy-2'-deoxyuridine, was found on the endothelial side of the cornea in addition to the parent compound; however, detectable levels of the metabolite have not been found in the aqueous humor in humans. It is unlikely that trifluridine is excreted in human milk after ophthalmic instillation of trifluridine because of the relatively small dosage (Metabolism / Metabolites Trifluridine is not metabolized by cytochrome P450 (CYP) enzymes. Trifluridine is mainly eliminated by metabolism via thymidine phosphorylase to form an inactive metabolite, 5-(trifluoromethyl) uracil (FTY). No other major metabolites were detected in plasma or urine. Other minor metabolites, such as 5-carboxy-2'-deoxyuridine found on the endothelial side of the cornea or 5-carboxyuraci, were also detected, but only at low or trace level in plasma and urine. The major metabolite of trifluridine (5-carboxy-2'-deoxyuridine appears) to have some antiviral activity but substantially less than that of the parent drug. 19F NMR spectroscopy has been used to study further the metabolism of 5-trifluoromethyl-2'-deoxyuridine (trifluridine; F3TdR). The synthesis and characterization of alpha-trifluoromethyl-beta-alanyl glycine (F3MBAG), a putative new metabolite of F3TdR, are now reported. This study describes ex vivo and in vivo detection of F3MBAG and other previously reported metabolites of trifluridine, using 19F NMR spectroscopy, in male BALB/C mice bearing EMT-6 tumors. A parallel 19F NMR spectroscopic study was also performed on rats dosed with F3TdR, to observe the qualitative pattern of F3TdR metabolism in another species. Unexpectedly, 5-trifluoromethyl-5,6-dihydroxyuracil (DOHF3T), alpha-trifluoromethyl-beta-ureidopropionic acid (F3MUPA) and fluoride, which result from the metabolic degradation of F3TdR and which were detected in various biological samples from mice dosed with F3TdR, could not be identified in rat urine or in homogenized tissue extracts. The presence of these metabolites in intact tissues is uncertain since in this study 19F NMR spectroscopy of these samples always displayed a broad resonance "hump" across the range of chemical shifts that would encompass these metabolites. No clear explanation for the loss of spectroscopic resolution in this region has been rationalized. N-Carboxy-alpha-trifluoromethyl-beta-alanine (F3MBA-CO2), alpha-trifluoromethyl-beta-alanyl alanine (F3MBAA) and N-acetyl-alpha-trifluoromethyl-beta-alanine (Ac-F3MBA) were synthesized and characterized, but were not detected as metabolites in any of the biological specimens examined. Biological Half-Life After administration of LONSURF 35 mg/m2, the mean elimination and steady-state half-life (t1/2) of trifluridine was 1.4 hours and 2.1 hours respectively. For the ophthalmic formulation, the half-life is significantly shorter, approximately only 12 minutes. |
||
| 毒性/毒理 (Toxicokinetics/TK) |
Hepatotoxicity
Pooled analyses of preregistration clinical trials reported that serum enzyme elevations occurred in up to 24% of patients on trifluridine/tipiracil therapy, but were also elevated in 27% of controls. Similarly, ALT values above 5 times ULN occurred in 2% of trifluridine/tipiracil treated compared to 4% of placebo treated subjects. In these and subsequent studies, clinically apparent liver adverse reactions attributed to trifluridine/tipiracil were not reported. Likelihood score: E (unlikely cause of clinically apparent liver injury). Protein Binding _In vitro_ findings suggest that the protein binding of trifluridine in human plasma is greater than 96%, where it is mainly bound to human serum albumin. Protein binding of trifluridine is independent of drug concentration and presence of tipiracil. |
||
| 参考文献 | |||
| 其他信息 |
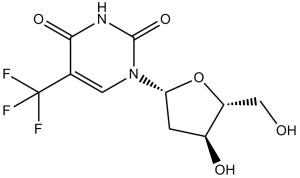
Trifluridine is a pyrimidine 2'-deoxyribonucleoside compound having 5-trifluoromethyluracil as the nucleobase. An antiviral drug used mainly in the treatment of primary keratoconjunctivitis and recurrent epithelial keratitis. It has a role as an antiviral drug, an antimetabolite, an EC 2.1.1.45 (thymidylate synthase) inhibitor and an antineoplastic agent. It is a nucleoside analogue, an organofluorine compound and a pyrimidine 2'-deoxyribonucleoside.
Trifluridine is a fluorinated pyrimidine nucleoside that is structurally related to [idoxuridine]. It is an active antiviral agent in ophthalmic solutions used mainly in the treatment of primary keratoconjunctivitis and recurrent epithelial keratitis due to herpes simplex virus. It displays effective antiviral activity against Herpes simplex virus type 1 and 2. The combination product of trifluridine with tipiracil marketed as Lonsurf has been approved in Japan, the United States, and the European Union for the treatment of adult patients with metastatic colorectal cancer who have been previously treated with fluoropyrimidine-, oxaliplatin- and irinotecan-based chemotherapy, an anti-VEGF biological therapy, and if RAS wild-type, an anti-EGFR therapy. In the anticancer therapy, trifluridine acts as a thymidine-based nucleoside metabolic inhibitor that gets incorporated into DNA of cancer cells following cell uptake to aberrate DNA function during cell replication. Trifluridine is a Nucleoside Analog Antiviral and Nucleoside Metabolic Inhibitor. The mechanism of action of trifluridine is as a Nucleic Acid Synthesis Inhibitor. Trifluridine/tipiracil is the combination of an antineoplastic pyrimidine analogue (trifluridine) with an inhibitor of its metabolism (tipiracil) that is used in the therapy of refractory, metastatic colorectal cancer. Trifluridine/tipiracil is associated with a low rate of transient serum enzyme elevations during therapy, but has not been implicated in cases of clinically apparent acute liver injury with jaundice. Trifluridine is a fluorinated thymidine analog with potential antineoplastic activity. Trifluridine is incorporated into DNA and inhibits thymidylate synthase, resulting in inhibition of DNA synthesis, inhibition of protein synthesis, and apoptosis. This agent also exhibits antiviral activity. (NCI04) An antiviral derivative of THYMIDINE used mainly in the treatment of primary keratoconjunctivitis and recurrent epithelial keratitis due to HERPES SIMPLEX virus. (From Martindale, The Extra Pharmacopoeia, 30th ed, p557) See also: Tipiracil hydrochloride; trifluridine (component of); Trifluridine; tipiracil (component of). Drug Indication As a standalone product, trifluridine is used for the ophthalmic treatment of primay keratoconjunctivitis and recurrent epithelial keratitis due to herpes simplex virus, types 1 and 2. Trifluridine is also available as a combination product with [tipiracil], which is indicated either alone or in combination with [bevacizumab] for the treatment of adult patients with metastatic colorectal cancer who have been previously treated with fluoropyrimidine-, oxaliplatin- and irinotecan-based chemotherapy, an anti-VEGF biological therapy, and if RAS wild-type, an anti-EGFR therapy. This combination product is also used for adult patients with metastatic gastric or gastroesophageal junction adenocarcinoma and were previously treated with at least two prior lines of chemotherapy that included a fluoropyrimidine, a platinum, either a taxane or irinotecan, and if appropriate, HER2/neu-targeted therapy. FDA Label Mechanism of Action The mechanism of action of trifluridine as an antiviral agent has not been fully elucidated, but appears to involve the inhibition of viral replication. Trifluridine gets incorporated into viral DNA during replication, which leads to the formation of defective proteins and an increased mutation rate. Trifluridine also mediates antineoplastic activities via this mechanism; following uptake into cancer cells, trifluridine is rapidly phosphorylated by thymidine kinase to its active monophosphate form. Subsequent phosphorylation produces trifluridine triphosphate, which is readily incorporated into the DNA of tumour cells in place of thymidine bases to perturb DNA function, DNA synthesis, and tumour cell proliferation. As trifluridine is subject to rapid degradation by TPase and readily metabolised by a first-pass effect following oral administration, tipiracil is added in the antineoplastic combination product as an inhibitor of TPase to increase the bioavailability of trifluridine. Trifluridine monophosphate also reversibly inhibits thymidylate synthetase (TS), an enzyme that is necessary for DNA synthesis and which levels are shown to be elevated different cancer cell lines. Up-regulation of the expression of the TS enzyme may also lead to the resistance to antineoplastic therapies, such as 5-fluorouracil (5-FU). [A35289 However, this inhibitory effect is not considered to be sufficient enough to fully contribute to the cytotoxicity in cancer cells. Trifluridine is a fluorinated pyrimidine nucleoside with in vitro and in vivo activity against herpes simplex virus, types 1 and 2 and vacciniavirus. Some strains of adenovirus are also inhibited in vitro. ...Trifluridine interferes with DNA synthesis in cultured mammalian cells. However, its antiviral mechanism of action is not completely known The exact mechanism of antiviral activity of trifluridine has not been fully elucidated, but appears to involve inhibition of viral replication. Trifluridine, instead of thymidine, is incorporated into viral DNA during replication which results in the formation of defective proteins and an increased mutation rate. Trifluridine also reversibly inhibits thymidylate synthetase, an enzyme required for DNA synthesis. Trifluridine has shown antiviral activity in vitro and in vivo against herpes simplex virus types 1 and 2 (HSV-1 and HSV-2). The drug is active in vitro against vaccinia virus and has shown in vivo activity in the treatment of vaccinia keratitis in rabbits. Trifluridine also has shown antiviral activity in cell culture against some strains of adenovirus. Trifluridine is inactive against bacteria, fungi, and Chlamydia. |
| 分子式 |
C10H11F3N2O5
|
|
|---|---|---|
| 分子量 |
296.2
|
|
| 精确质量 |
296.062
|
|
| 元素分析 |
C, 40.55; H, 3.74; F, 19.24; N, 9.46; O, 27.01
|
|
| CAS号 |
70-00-8
|
|
| 相关CAS号 |
|
|
| PubChem CID |
6256
|
|
| 外观&性状 |
White to off-white solid powder
|
|
| 密度 |
1.6±0.1 g/cm3
|
|
| 熔点 |
190-193 °C(lit.)
|
|
| 折射率 |
1.534
|
|
| LogP |
0.07
|
|
| tPSA |
104.55
|
|
| 氢键供体(HBD)数目 |
3
|
|
| 氢键受体(HBA)数目 |
8
|
|
| 可旋转键数目(RBC) |
2
|
|
| 重原子数目 |
20
|
|
| 分子复杂度/Complexity |
464
|
|
| 定义原子立体中心数目 |
3
|
|
| SMILES |
FC(C1C(N([H])C(N(C=1[H])[C@@]1([H])C([H])([H])[C@@]([H])([C@@]([H])(C([H])([H])O[H])O1)O[H])=O)=O)(F)F
|
|
| InChi Key |
VSQQQLOSPVPRAZ-RRKCRQDMSA-N
|
|
| InChi Code |
InChI=1S/C10H11F3N2O5/c11-10(12,13)4-2-15(9(19)14-8(4)18)7-1-5(17)6(3-16)20-7/h2,5-7,16-17H,1,3H2,(H,14,18,19)/t5-,6+,7+/m0/s1
|
|
| 化学名 |
1-[(2R,4S,5R)-4-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-5-(trifluoromethyl)pyrimidine-2,4-dione
|
|
| 别名 |
|
|
| HS Tariff Code |
2934.99.9001
|
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
|
|||
|---|---|---|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 2.5 mg/mL (8.44 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 25.0 mg/mL澄清DMSO储备液加入到400 μL PEG300中,混匀;然后向上述溶液中加入50 μL Tween-80,混匀;加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: ≥ 2.5 mg/mL (8.44 mM) (饱和度未知) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 25.0 mg/mL澄清DMSO储备液加入900 μL 20% SBE-β-CD生理盐水溶液中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 View More
配方 3 中的溶解度: ≥ 2.5 mg/mL (8.44 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 3.3761 mL | 16.8805 mL | 33.7610 mL | |
| 5 mM | 0.6752 mL | 3.3761 mL | 6.7522 mL | |
| 10 mM | 0.3376 mL | 1.6880 mL | 3.3761 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
| NCT Number | Recruitment | interventions | Conditions | Sponsor/Collaborators | Start Date | Phases |
| NCT04737187 | Active Recruiting |
Drug: Trifluridine/Tipiracil Drug: Bevacizumab |
Refractory Metastatic Colorectal Cancer |
Taiho Oncology, Inc. | November 25, 2020 | Phase 3 |
| NCT03992456 | Active Recruiting |
Biological: Panitumumab Drug: Regorafenib |
Metastatic Colorectal Carcinoma Stage III Colon Cancer AJCC v8 |
Academic and Community Cancer Research United |
April 24, 2020 | Phase 2 |
| NCT03981614 | Active Recruiting |
Drug: Palbociclib Drug: Binimetinib |
Unresectable Carcinoma Metastatic Colorectal Carcinoma |
Academic and Community Cancer Research United |
October 29, 2019 | Phase 2 |
| NCT05198934 | Active Recruiting |
Drug: Trifluridine and Tipiracil Drug: Regorafenib |
Colorectal Cancer (CRC) | Amgen | April 19, 2022 | Phase 3 |
| NCT05608044 | Active Recruiting |
Drug: Botensilimab Drug: Balstilimab |
Metastatic Colorectal Cancer | Agenus Inc. | November 30, 2022 | Phase 2 |
|
|
 RNase L-IN-2
RNase L-IN-2
 BW 348U87
BW 348U87
 ERCC1/XPA interaction inhibitor 1
ERCC1/XPA interaction inhibitor 1
 PHYLPA-8
PHYLPA-8
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1





























 COA
COA


