| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 10 mM * 1 mL in DMSO |
|
||
| 1mg |
|
||
| 2mg |
|
||
| 5mg |
|
||
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| 500mg |
|
||
| 1g |
|
||
| Other Sizes |
|

| 靶点 |
WT ALK (IC50 = 1.01 nM); ALK(L1196M) (IC50 = 1.08 nM); ALK(G1202R) (IC50 = 1.26 nM); Trk receptor; ROS1
|
||
|---|---|---|---|
| 体外研究 (In Vitro) |
体外活性:TPX-0005 是一种口服有效的 ATP 竞争性抑制剂,针对 ALK、ROS1、TRKA、TRKB 和 TRKC 重组激酶及其相应的临床耐药突变体。TPX-0005 在伤口愈合试验中抑制 H2228 细胞迁移,效果类似萨拉卡替尼的活性。它不仅可以抑制野生型和广谱突变型 ALK,还可以通过抑制 SRC 克服原发性耐药并抑制转移特征。激酶测定:TPX-0005 是一种新型、合理设计的高效 ALK/ROS1/TRK 抑制剂,对 SRC、WT ALK、ALK G1202R 和 ALK L1196M 的 IC50 分别为 5.3 nM、1.01 nM、1.26 nM 和 1.08 nM 。它具有潜在的抗癌活性。它通过强烈抑制 EML4-ALK (IC50 13 nM) 和 SRC 底物桩蛋白 (IC50 107 nM) 的磷酸化,有效克服了这种主要耐药性(细胞增殖测定中 IC50 100 nM)。 PX-0005 在伤口愈合试验中抑制 H2228 细胞迁移,其活性与萨拉卡替尼相似。总体而言,TPX-0005 具有非常有利的特性,能够克服多种 ALK 耐药机制,包括二次突变、旁路信号激活、EMT,并值得临床研究细胞测定:TPX-0005 也是一种有效的 SRC 抑制剂 (IC50 5.3 nM)。 H2228 肺癌细胞系中 SRC 激酶活性升高,导致细胞增殖测定中对克唑替尼 (IC50 1200 nM) 和色瑞替尼 (IC50 1000 nM) 产生耐药性。 TPX-0005 通过强烈抑制 EML4-ALK (IC50 13 nM) 和 SRC 底物桩蛋白 (IC50 107 nM) 以及其他下游信号传导靶标的磷酸化,有效克服了这种主要耐药性(细胞增殖测定中 IC50 100 nM)。 TPX-0005 在伤口愈合试验中抑制 H2228 细胞迁移,其活性与萨拉卡替尼相似。
|
||
| 体内研究 (In Vivo) |
在患者来源的异种移植肿瘤模型中,TPX-0005 治疗可导致含有致癌 ALK、ROS1 和 TRKC 融合的肿瘤显着消退。此外,在一系列小鼠异种移植肿瘤模型中,TPX-0005不仅在含有野生型致癌靶点的肿瘤中表现出显着的抗肿瘤活性,而且通过抑制靶点磷酸化,在含有溶剂前沿突变的癌基因的肿瘤中也表现出显着的抗肿瘤活性。
|
||
| 酶活实验 |
TPX-0005 是一种新型、合理设计的高效 ALK/ROS1/TRK 抑制剂,对 SRC、WT ALK、ALK G1202R 和 ALK L1196M 的 IC50 值为 5.3 nM、1.01 nM、1.26 nM 和 1.08 nM。那个订单。它可能具有抗癌特性。通过显着抑制 EML4-ALK (IC50 13 nM) 和 SRC 底物桩蛋白 (IC50 107 nM) 的磷酸化,它成功克服了这种主要耐药性(细胞增殖测定中 IC50 100 nM)。在伤口愈合试验中,PX-0005 抑制 H2228 细胞迁移,其活性与 saracatinib 相当。综合考虑,TPX-0005 具有非常有前途的特性,能够克服多种 ALK 耐药机制,例如二次突变、旁路信号激活和 EMT。因此,值得进一步的临床研究。
|
||
| 细胞实验 |
TPX-0005 也是一种有效的 SRC 抑制剂 (IC50 5.3 nM)。在细胞增殖测试中,H2228 肺癌细胞系中 SRC 激酶活性的增加导致对克唑替尼 (IC50 1200 nM) 和色瑞替尼 (IC50 1000 nM) 的耐药性。通过强烈抑制 EML4-ALK (IC50 13 nM) 和 SRC 底物桩蛋白 (IC50 107 nM) 以及其他下游信号传导靶标的磷酸化,TPX-0005 有效克服了这种主要耐药性(细胞增殖测定中 IC50 100 nM) )。与 saracatinib 类似,TPX-0005 在伤口愈合试验中抑制 H2228 细胞迁移。
|
||
| 动物实验 |
|
||
| 药代性质 (ADME/PK) |
Absorption, Distribution and Excretion
The geometric mean (CV%) of repotrectinib steady state peak concentration (Cmax,ss) is 713 (32.5%) ng/mL and the area under the time concentration curve (AUC0-24h,ss) is 7210 (40.1%) ng•h/mL following the approved recommended twice daily dosage in patients with cancer. Repotrectinib Cmax and AUC0-inf increases approximately dose-proportional (but less than linear with estimated slopes of 0.78 and 0.70, respectively) over the single dose range of 40 mg to 240 mg (0.25 to 1.5 times the approved recommended dosage). Steady-state PK was time-dependent with an autoinduction of CYP3A4. Steady-state is achieved within 14 days of daily administration of 160 mg. The geometric mean (CV%) absolute bioavailability of repotrectinib is 45.7% (19.6%). Peak repotrectinib concentration occurred at approximately 2 to 3 hours post a single oral dose of 40 mg to 240 mg (0.25 to 1.5 times the approved recommended dosage) under fasted conditions. No clinically significant differences in repotrectinib pharmacokinetics were observed in patients with cancer following administration of a high-fat meal (approximately 800-1000 calories, 50% fat). Following a single oral 160 mg dose of radiolabeled repotrectinib, 4.84% (0.56% as unchanged) was recovered in urine and 88.8% (50.6% unchanged) in feces. The geometric mean (CV%) apparent volume of distribution (Vz/F) was 432 L (55.9%) in patients with cancer following a single 160 mg oral dose of repotrectinib. The geometric mean (CV%) apparent oral clearance (CL/F) was 15.9 L/h (45.5%) in patients with cancer following a single 160 mg oral dose of repotrectinib. Metabolism / Metabolites Repotrectinib is primarily metabolized by CYP3A4 followed by secondary glucuronidation. Biological Half-Life The repotrectinib mean terminal half-life is approximately 50.6 h for patients with cancer following a single dose. The steady-state repotrectinib terminal half-life is approximately 35.4 h for patients with cancer. |
||
| 毒性/毒理 (Toxicokinetics/TK) |
Effects During Pregnancy and Lactation
◉ Summary of Use during Lactation No information is available on the use of repotrectinib during breastfeeding. Because repotrectinib is 94.5% bound to plasma proteins, the amount in milk is likely to be low and oral bioavailability is less than 50%; however, the drug’s half-life is about 50 hours in adults. The manufacturer recommends that breastfeeding be discontinued during repotrectinib therapy and for 10 days after the final dose. ◉ Effects in Breastfed Infants Relevant published information was not found as of the revision date. ◉ Effects on Lactation and Breastmilk Relevant published information was not found as of the revision date. Protein Binding Repotrectinib binding to plasma protein was 95.4% in vitro. The blood-to-plasma ratio was 0.56 in vitro. |
||
| 参考文献 | |||
| 其他信息 |
Repotrectinib is an azamacrocycle with formula C18H18FN5O2. It is a tyrosine kinase inhibitor (highly potent against ROS1, TRKA-C, and ALK) used for the treatment of locally advanced or metastatic ROS1-positive non-small cell lung cancer. It has a role as an EC 2.7.10.1 (receptor protein-tyrosine kinase) inhibitor and an antineoplastic agent. It is a member of monofluorobenzenes, a pyrazolopyrimidine, a cyclic ether, a secondary carboxamide and an azamacrocycle.
Repotrectinib is a next-generation tyrosine kinase inhibitor (TKI) specifically designed to address resistance in the treatment of non-small cell lung cancer (NSCLC), specifically due to mutations in the ROS1 gene. ROS1 mutations are one of the defined oncogenic drives of NSCLC, and the solvent-front mutation ROS1 G2032R is responsible for 50 to 60% of [crizotinib]-resistant cases. Repotrectinib possesses a compact macrocyclic structure that both limits adverse interactions with resistance mutation hotspots and targets mutations in the solvent-front region. Although resistance to multiple TKI has been reported, including [crizotinib], [lorlatinib], [taletrectinib], and [entrectinib], there has been no reported case of repotrectinib resistance. On November 15th, 2023, the FDA approved repotrectinib under the brand name Augtyro for the treatment of locally advanced or metastatic ROS1-Positive NSCLC. This approval is based on favorable results from the TRIDENT-1 study, where the objective response rate was 79% in TKI-naive patients and 38% in TKI-pretreated patients respectively. Repotrectinib is a Kinase Inhibitor. The mechanism of action of repotrectinib is as a Proto-Oncogene Tyrosine-Protein Kinase ROS1 Inhibitor, and Tropomyosin Receptor Tyrosine Kinase A Inhibitor, and Tropomyosin Receptor Tyrosine Kinase B Inhibitor, and Tropomyosin Receptor Tyrosine Kinase C Inhibitor, and Cytochrome P450 3A4 Inducer. Repotrectinib is an orally available inhibitor of multiple kinases, including the receptor tyrosine kinase anaplastic lymphoma kinase (ALK), c-ros oncogene 1 (ROS1), the neurotrophic tyrosine receptor kinase (NTRK) types 1, 2 and 3, the proto-oncogene SRC, and focal adhesion kinase (FAK), with potential antineoplastic activity. Upon oral administration, repotrectinib binds to and inhibits wild-type, point mutants and fusion proteins of ALK, ROS1, NTRK1-3, SRC, FAK and, to a lesser extent, other kinases. Inhibition of these kinases leads to the disruption of downstream signaling pathways and the inhibition of cell growth of tumors in which these kinases are overexpressed, rearranged or mutated. Drug Indication Repotrectinib is indicated for the treatment of adult patients with locally advanced or metastatic ROS1-positive non-small cell lung cancer (NSCLC). Treatment of all conditions included in the category of malignant neoplasms (except haematopoietic neoplasms) Mechanism of Action Repotrectinib is an inhibitor of proto-oncogene tyrosine-protein kinase ROS1 (ROS1) and of the tropomyosin receptor tyrosine kinases (TRKs) TRKA, TRKB, and TRKC. |
| 分子式 |
C18H18FN5O2
|
|---|---|
| 分子量 |
355.37
|
| 精确质量 |
355.144
|
| 元素分析 |
C, 60.84; H, 5.11; F, 5.35; N, 19.71; O, 9.00
|
| CAS号 |
1802220-02-5
|
| 相关CAS号 |
1802220-02-5; 2058227-19-1
|
| PubChem CID |
135565923
|
| 外观&性状 |
White to off-white solid powder
|
| 密度 |
1.5±0.1 g/cm3
|
| 折射率 |
1.694
|
| LogP |
1.71
|
| tPSA |
80.6
|
| 氢键供体(HBD)数目 |
2
|
| 氢键受体(HBA)数目 |
6
|
| 可旋转键数目(RBC) |
0
|
| 重原子数目 |
26
|
| 分子复杂度/Complexity |
524
|
| 定义原子立体中心数目 |
2
|
| SMILES |
FC1C=CC2=C(C=1)[C@@H](C)NC1C=CN3C(=C(C=N3)C(NC[C@H](C)O2)=O)N=1
|
| InChi Key |
FIKPXCOQUIZNHB-WDEREUQCSA-N
|
| InChi Code |
InChI=1S/C18H18FN5O2/c1-10-8-20-18(25)14-9-21-24-6-5-16(23-17(14)24)22-11(2)13-7-12(19)3-4-15(13)26-10/h3-7,9-11H,8H2,1-2H3,(H,20,25)(H,22,23)/t10-,11+/m0/s1
|
| 化学名 |
(3R,11S)-6-fluoro-3,11-dimethyl-10-oxa-2,13,17,18,21-pentazatetracyclo[13.5.2.04,9.018,22]docosa-1(21),4(9),5,7,15(22),16,19-heptaen-14-one
|
| 别名 |
Ropotrectinib; TPX0005; TPX-0005; TPX 0005; Augtyro
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
|
|||
|---|---|---|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 2.5 mg/mL (7.03 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 25.0 mg/mL澄清DMSO储备液加入到400 μL PEG300中,混匀;然后向上述溶液中加入50 μL Tween-80,混匀;加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: ≥ 2.5 mg/mL (7.03 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 25.0 mg/mL 澄清 DMSO 储备液加入到 900 μL 玉米油中并混合均匀。 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.8140 mL | 14.0698 mL | 28.1397 mL | |
| 5 mM | 0.5628 mL | 2.8140 mL | 5.6279 mL | |
| 10 mM | 0.2814 mL | 1.4070 mL | 2.8140 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
| NCT Number | Recruitment | interventions | Conditions | Sponsor/Collaborators | Start Date | Phases |
| NCT04094610 | Recruiting | Drug: Oral repotrectinib (TPX-0005) |
Lymphoma Primary CNS Tumors |
Turning Point Therapeutics, Inc. | March 12, 2020 | Phase 1 Phase 2 |
| NCT05004116 | Recruiting | Drug: Irinotecan and temozolomide Drug: Repotrectinib |
Advanced Cancer Metastatic Solid Tumor |
Memorial Sloan Kettering Cancer Center |
August 9, 2021 | Phase 1 Phase 2 |
| NCT04772235 | Recruiting | Drug: Repotrectinib Drug: Osimertinib |
Nsclc | Instituto Oncológico Dr Rosell | February 11, 2022 | Phase 1 |
| NCT05828303 | Recruiting | Drug: TPX-0005 Drug: Digoxin |
Advanced Solid Tumor Metastatic Solid Tumor |
Turning Point Therapeutics, Inc. | July 28, 2022 | Phase 1 |
| NCT03093116 | Recruiting | Drug: Oral repotrectinib (TPX-0005) |
Locally Advanced Solid Tumors Metastatic Solid Tumors |
Turning Point Therapeutics, Inc. | February 27, 2017 | Phase 1 Phase 2 |
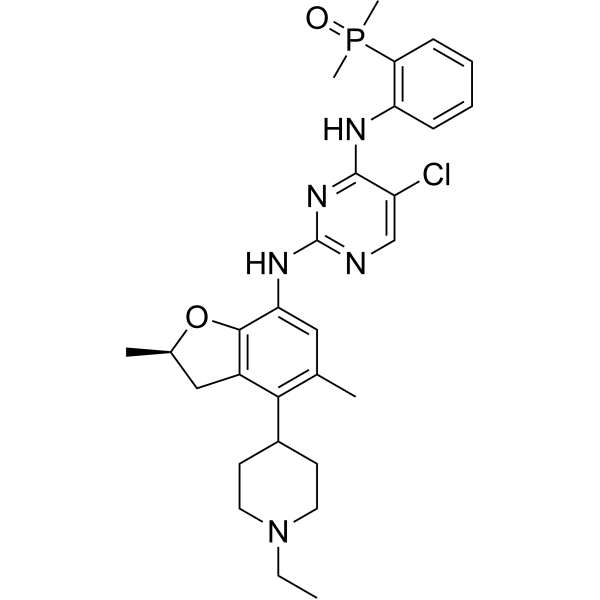 ROS1-IN-2
ROS1-IN-2
 PRDX1-IN-1
PRDX1-IN-1
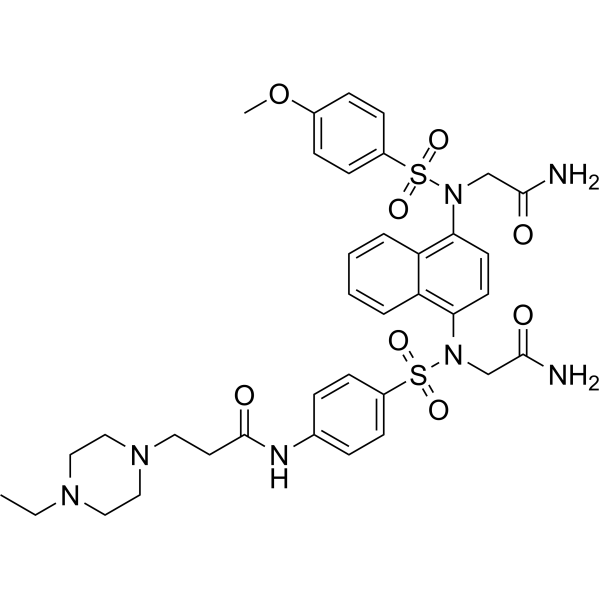 Keap1-Nrf2-IN-11
Keap1-Nrf2-IN-11
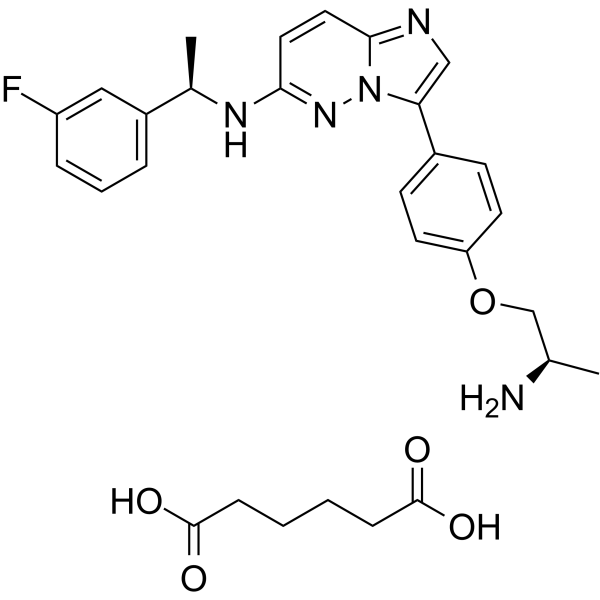 Taletrectinib adipate (DS-6051b; AB-106; IBI-344)
Taletrectinib adipate (DS-6051b; AB-106; IBI-344)
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1





























 COA
COA
