| 规格 | 价格 | |
|---|---|---|
| 500mg | ||
| 1g | ||
| Other Sizes |
| 靶点 |
MNK1 (IC50 = 1-2 nM); MNK2 (IC50 = 1-2 nM); PD-L1
|
||
|---|---|---|---|
| 体外研究 (In Vitro) |
Tomivosertib (eFT508) 以剂量依赖性方式降低肿瘤细胞系中丝氨酸 209 处的 eIF4E 磷酸化 (IC50 = 2–16 nM)。 Tomivosertib 对大约 50 种血液癌症中的多种 DLBCL 细胞系表现出抗增殖活性。 TMD8、OCI-Ly3 和 HBL1 DLBCL 细胞系对 tomivosertib 的敏感性与促炎细胞因子(如 TNF、IL-6、IL-10 和 CXCL10)产生的剂量依赖性减少有关。对 Tomivosertib 作用模式的更彻底分析表明,TNF 合成减少与 TNFα mRNA 半衰期缩短 2 倍相关[1]。
|
||
| 体内研究 (In Vivo) |
Tomivosertib (eFT508) 在 TMD8 和 HBL-1 ABC-DLBCL 模型中表现出显着的抗肿瘤活性,这两种模型都含有激活的 MyD88 突变。此外,在人类淋巴瘤模型中,tomovosertib 与 R-CHOP 成分以及 PCI-32765 和 Venetoclax 等全新靶向药物有效相互作用[1]。
|
||
| 酶活实验 |
在许多实体瘤和血液恶性肿瘤的发病机制中,信使 RNA (mRNA) 翻译失调。 MNK1 和 MNK2 磷酸化真核起始因子 4E (eIF4E) 和其他重要效应蛋白(如 hnRNPA1 和 PSF),以整合来自各种免疫和致癌信号通路(如 RAS、p38 和 Toll 样受体 (TLR) 通路)的信号。 MNK1 和 MNK2 通过这些调节蛋白的磷酸化专门控制细胞 mRNA 的稳定性和翻译子集。 eFT508 是一种强效、选择性极高、可口服生物利用的 MNK1 和 MNK2 抑制剂。在酶测定中,eFT508 通过 ATP 竞争性可逆机制抑制激酶,对两种 MNK 同工型的半数抑制浓度 (IC50) 为 1-2 nM。
|
||
| 细胞实验 |
用eFT508治疗肿瘤细胞系导致eIF4E丝氨酸209位点磷酸化的剂量依赖性降低(IC50 = 2-16 nM),这与先前的研究结果一致,即该位点的磷酸化仅依赖于MNK1/MNK2。在约50例血液肿瘤中,eFT508对多种DLBCL细胞系显示出抗增殖活性。TMD8、OCI-Ly3和HBL1 DLBCL细胞系对eFT508的敏感性与促炎细胞因子(包括TNFα、IL-6、IL-10和CXCL10)的产生呈剂量依赖性减少有关。进一步评估eFT508的作用机制表明,TNFα产生的减少与TNFα mRNA半衰期减少2倍相关。[1]
荧光素酶检测。[2] KRASG12D和MYCTg;KRASG12D细胞在12孔板中转染200 ng pGL3 (Firelfy荧光素酶)构建物,其中含有PD-L1全长或突变的5'UTR和40 ng pRL (Renilla荧光素酶)质粒,使用Lipofectamine 2000根据制造商的说明。转染24 h后收集细胞,一半细胞用Dual luciferase kit检测,另一半细胞用TRIzol纯化RNA。将萤火虫的荧光素酶活性归一化为鼠兔的活性,并进一步归一化为萤火虫和鼠兔荧光素酶RNA的RT-qPCR定量。 将 eFT508 按建议浓度应用于 TMD8 细胞 24 小时。 m7-GTP 用于细胞裂解物。免疫印迹用于检查琼脂糖凝胶拉下的蛋白质和结合的蛋白质。 |
||
| 动物实验 |
|
||
| 药代性质 (ADME/PK) |
Results for clinical and pharmacokinetic endpoints
Participant characteristics are summarized in Supplementary Tables S1 and S2. Nineteen patients with metastatic breast cancer ranging in age from 27 to 77 were enrolled. The study population included patients with estrogen receptor–positive (ER+), Her2-positive, and triple-negative types of breast cancer. To be eligible, participants had to have progressive disease on approved therapies or refused approved therapies. As shown in Supplementary Table S3, the majority were heavily pretreated for metastatic breast cancer. Safety was evaluated during the 2-week run-in period in which tomivosertib was given as a single agent and during subsequent treatment in which tomivosertib was given in combination with paclitaxel. As shown in Supplementary Tables S4A, S4B, S5, and S6, no patient stopped treatment for adverse events related to tomivosertib toxicity. Physician-determined adverse reactions related to tomivosertib were mostly low-grade changes in serum biochemistry results, including liver enzymes. Pharmacokinetic studies were undertaken in 12 patients, and results are shown in Supplementary Fig. S2A and S2B. As expected, serum paclitaxel was undetectable during the tomivosertib-alone run-in period and peaked at the end of infusion at ∼2,200 ng/mL followed by a rapid drop to ∼ 400 ng/mL 1 hour after the end of infusion and a steady decline over the subsequent 36 hours. When paclitaxel was administered, patients were already receiving oral tomivosertib, and the presence of tomivosertib was noted to have no major effect on paclitaxel levels previously observed following infusion as a single agent. This finding is consistent with the absence of observed increase in paclitaxel toxicity in the presence of tomivosertib. Tomivosertib serum levels following oral dosing at 100 mg orally twice daily exhibited some variability between patients, but the minimum concentration remained above 98 ng/mL in three patients who took the drug at 100 mg twice daily without meals, whereas the minimum concentration was above 156 ng/mL in nine patients who took the drug at this dose with meals. Importantly, there was no major change in tomivosertib concentration whether paclitaxel was present or not. The measured serum concentrations were in a range previously observed to have in vitro activity. As a phase Ib study, the study was designed primarily to provide information about safety, pharmacokinetics, and pharmacodynamics. No conclusions can be drawn about clinical utility in view of the absence of a control group and the small number of patients. As shown in Fig. 2, one patient had stable disease for 13 months and two others had stable disease for 8 months while receiving treatment according to the protocol. https://pubmed.ncbi.nlm.nih.gov/39576211/ |
||
| 参考文献 |
|
||
| 其他信息 |
Tomivosertib is under investigation in clinical trial NCT03318562 (A PD Study of Oral eFT508 in Subjects With Advanced TNBC and HCC).
Tomivosertib is an orally bioavailable inhibitor of mitogen-activated protein kinase (MAPK)-interacting serine/threonine-protein kinase 1 (MNK1) and 2 (MNK2), with potential antineoplastic activity. Upon oral administration, tomivosertib binds to and inhibits the activity of MNK1 and 2. This prevents MNK1/2-mediated signaling, and inhibits the phosphorylation of certain regulatory proteins, including eukaryotic translation initiation factor 4E (eIF4E), that regulate the translation of messenger RNAs (mRNAs) involved in tumor cell proliferation, angiogenesis, survival and immune signaling. This inhibits tumor cell proliferation in MNK1/2-overexpressing tumor cells. MNK1/2 are overexpressed in a variety of tumor cell types and promote phosphorylation of eIF4E; eIF4E is overexpressed in many tumor cell types and contributes to tumor development, maintenance and resistance. Dysregulated translation of messenger RNA (mRNA) plays a role in the pathogenesis of multiple solid tumors and hematological malignancies. MNK1 and MNK2 integrate signals from several oncogenic and immune signaling pathways, including RAS, p38, and Toll-like receptor (TLR) pathways, by phosphorylating eukaryotic initiation factor 4E (eIF4E) and other key effector proteins including hnRNPA1 and PSF. Through phosphorylation of these regulatory proteins MNK1 and MNK2 selectively regulate the stability and translation of a subset of cellular mRNA. eFT508 is a potent, highly selective, and orally bioavailable MNK1 and MNK2 inhibitor. eFT508 has a half-maximal inhibitory concentration (IC50) of 1-2 nM against both MNK isoforms in enzyme assays and inhibits the kinase through a reversible, ATP-competitive mechanism of action. Treatment of tumor cell lines with eFT508 led to a dose-dependent reduction in eIF4E phosphorylation at serine 209 (IC50 = 2-16 nM), consistent with previous findings that phosphorylation of this site is solely dependent upon MNK1/MNK2. In a panel of ~50 hematological cancers, eFT508 showed anti-proliferative activity against multiple DLBCL cell lines. Sensitivity to eFT508 in TMD8, OCI-Ly3 and HBL1 DLBCL cell lines was associated with dose-dependent decreases in production of pro-inflammatory cytokines including TNFα, IL-6, IL-10 and CXCL10. Further evaluation eFT508 mechanism of action demonstrated that decreased TNFα production correlated with a 2-fold decrease in TNFα mRNA half-life. These findings are consistent with MNK1 phosphorylation of specific RNA-binding proteins, eg, hnRNPA1, that regulate the stability and translation of mRNA containing specific AU-rich elements (ARE) in their 3'-untranslated regions (UTR). Pro-inflammatory cytokines are drivers of key hallmarks of cancer including tumor cell survival, migration and invasion, angiogenesis, and immune evasion, while also driving drug resistance. Therefore, eFT508 was tested in vivo in 7 subcutaneous human lymphoma xenograft models. Significant anti-tumor activity was observed in the TMD8 and HBL-1 ABC-DLBCL models, both of which harbor activating MyD88 mutations. In addition, eFT508 combined effectively with components of R-CHOP and with novel targeted agents, including ibrutinib and venetoclax, in human lymphoma models. These results underscore the potential of eFT508 for the treatment of DLBCL. eFT508 has also been characterized in nonclinical safety pharmacology and toxicology studies. Clinical trials in patients with hematological and other malignancies are planned.[1] Cancer cells develop mechanisms to escape immunosurveillance, among which modulating the expression of immune suppressive messenger RNAs is most well-documented. However, how this is molecularly achieved remains largely unresolved. Here, we develop an in vivo mouse model of liver cancer to study oncogene cooperation in immunosurveillance. We show that MYC overexpression (MYCTg) synergizes with KRASG12D to induce an aggressive liver tumor leading to metastasis formation and reduced mouse survival compared with KRASG12D alone. Genome-wide ribosomal footprinting of MYCTg;KRASG12 tumors compared with KRASG12D revealed potential alterations in translation of mRNAs, including programmed-death-ligand 1 (PD-L1). Further analysis revealed that PD-L1 translation is repressed in KRASG12D tumors by functional, non-canonical upstream open reading frames in its 5' untranslated region, which is bypassed in MYCTg;KRASG12D tumors to evade immune attack. We show that this mechanism of PD-L1 translational upregulation was effectively targeted by a potent, clinical compound that inhibits eIF4E phosphorylation, eFT508, which reverses the aggressive and metastatic characteristics of MYCTg;KRASG12D tumors. Together, these studies reveal how immune-checkpoint proteins are manipulated by distinct oncogenes at the level of mRNA translation, which can be exploited for new immunotherapies.[2] |
| 分子式 |
C17H21CLN6O2
|
|---|---|
| 分子量 |
376.840641736984
|
| 精确质量 |
376.141
|
| 元素分析 |
C, 54.18; H, 5.62; Cl, 9.41; N, 22.30; O, 8.49
|
| CAS号 |
1849590-02-8
|
| 相关CAS号 |
1849590-02-8 (HCl);1849590-01-7;
|
| PubChem CID |
118598855
|
| 外观&性状 |
Typically exists as solid at room temperature
|
| tPSA |
113
|
| 氢键供体(HBD)数目 |
4
|
| 氢键受体(HBA)数目 |
6
|
| 可旋转键数目(RBC) |
2
|
| 重原子数目 |
26
|
| 分子复杂度/Complexity |
664
|
| 定义原子立体中心数目 |
0
|
| InChi Key |
WBGPPUUXCGKTSC-UHFFFAOYSA-N
|
| InChi Code |
InChI=1S/C17H20N6O2.ClH/c1-10-7-11(21-13-8-12(18)19-9-20-13)16(25)23-14(10)15(24)22-17(23)5-3-2-4-6-17/h7-9H,2-6H2,1H3,(H,22,24)(H3,18,19,20,21)1H
|
| 化学名 |
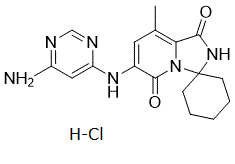
6'-((6-aminopyrimidin-4-yl)amino)-8'-methyl-2'H-spiro[cyclohexane-1,3'-imidazo[1,5-a]pyridine]-1',5'-dione
hydrochloride
|
| 别名 |
Tomivosertib HCl; eFT508; eFT-508; eFT508HCl; Tomivosertib hydrochloride; EFT-508 hydrochloride; Tomivosertib (hydrochloride); BW3S40K2UM; Tomivosertib hydrochloride [USAN]; eFT508 HCl; Tomivosertib HCl; eFT 508; eFT508 hydrochloride
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples
|
|---|---|
| 溶解度 (体内实验) |
注意: 如下所列的是一些常用的体内动物实验溶解配方,主要用于溶解难溶或不溶于水的产品(水溶度<1 mg/mL)。 建议您先取少量样品进行尝试,如该配方可行,再根据实验需求增加样品量。
注射用配方
注射用配方1: DMSO : Tween 80: Saline = 10 : 5 : 85 (如: 100 μL DMSO → 50 μL Tween 80 → 850 μL Saline)(IP/IV/IM/SC等) *生理盐水/Saline的制备:将0.9g氯化钠/NaCl溶解在100 mL ddH ₂ O中,得到澄清溶液。 注射用配方 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (如: 100 μL DMSO → 400 μL PEG300 → 50 μL Tween 80 → 450 μL Saline) 注射用配方 3: DMSO : Corn oil = 10 : 90 (如: 100 μL DMSO → 900 μL Corn oil) 示例: 以注射用配方 3 (DMSO : Corn oil = 10 : 90) 为例说明, 如果要配制 1 mL 2.5 mg/mL的工作液, 您可以取 100 μL 25 mg/mL 澄清的 DMSO 储备液,加到 900 μL Corn oil/玉米油中, 混合均匀。 View More
注射用配方 4: DMSO : 20% SBE-β-CD in Saline = 10 : 90 [如:100 μL DMSO → 900 μL (20% SBE-β-CD in Saline)] 口服配方
口服配方 1: 悬浮于0.5% CMC Na (羧甲基纤维素钠) 口服配方 2: 悬浮于0.5% Carboxymethyl cellulose (羧甲基纤维素) 示例: 以口服配方 1 (悬浮于 0.5% CMC Na)为例说明, 如果要配制 100 mL 2.5 mg/mL 的工作液, 您可以先取0.5g CMC Na并将其溶解于100mL ddH2O中,得到0.5%CMC-Na澄清溶液;然后将250 mg待测化合物加到100 mL前述 0.5%CMC Na溶液中,得到悬浮液。 View More
口服配方 3: 溶解于 PEG400 (聚乙二醇400) 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.6536 mL | 13.2682 mL | 26.5365 mL | |
| 5 mM | 0.5307 mL | 2.6536 mL | 5.3073 mL | |
| 10 mM | 0.2654 mL | 1.3268 mL | 2.6536 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
| NCT Number | Recruitment | interventions | Conditions | Sponsor/Collaborators | Start Date | Phases |
| NCT05744739 | Recruiting | Procedure: Biospecimen Collection Drug: Tomivosertib |
Acute Myeloid Leukemia | Northwestern University | September 29, 2023 | Phase 1 |
| NCT04622007 | Recruiting | Drug: Tomivosertib Drug: Pemetrexed |
Non-small Cell Lung Cancer | Effector Therapeutics | June 2, 2021 | Phase 2 |
| NCT04261218 | Completed | Drug: tomivosertib Drug: paclitaxel |
Breast Cancer | Translational Research in Oncology |
August 25, 2020 | Phase 2 |
| NCT03616834 | Completed | Drug: Tomivosertib (eFT-508) |
Solid Tumors | Effector Therapeutics | July 25, 2018 | Phase 2 |
| NCT02937675 | Terminated | Drug: Tomivosertib (eFT-508) |
Lymphoma | Effector Therapeutics | February 8, 2017 | Phase 1 Phase 2 |
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1






























 COA
COA




