
| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 10 mM * 1 mL in DMSO |
|
||
| 1mg |
|
||
| 5mg |
|
||
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| 500mg |
|
||
| 1g |
|
||
| Other Sizes |
|
| 靶点 |
vasopressin receptor 2 ( IC50 = 3 nM )
|
|---|---|
| 体外研究 (In Vitro) |
体外活性:托伐普坦阻断 [(3)H]AVP 与人 V(2) 受体的结合,选择性比 V(1a) 受体高 29 倍,并且对 V(1b) 受体没有抑制作用。 Tolvaptan 不仅能抑制 [(3)H]AVP 的结合,还能抑制表达人 V(2) 受体的 HeLa 细胞中 AVP 诱导的环 AMP 的产生。托伐普坦在健康和患病动物中表现出明显的渗水现象。 Tolvaptan 在常染色体显性多囊肾病 (ADPKD) 细胞中对精氨酸加压素诱导的 cAMP 产生产生浓度依赖性抑制,表观 IC(50) 为 0.1 nM。 Tolvaptan 抑制 AVP 诱导的 ERK 信号传导和细胞增殖。 Tolvaptan 抑制 AVP 诱导的 Cl(-) 分泌,并减少在三维胶原基质中培养的 ADPKD 细胞的体外囊肿生长。
|
| 体内研究 (In Vivo) |
托伐普坦可改善低钠血症,从而预防死亡,并改善急性和慢性低钠血症大鼠模型的器官水潴留。托伐普坦可降低心脏前负荷,且不会对心力衰竭 (HF) 犬的肾功能、全身血流动力学或循环神经激素产生不利影响。在人类多囊肾病 (PKD) 动物模型中,托伐普坦显示肾脏重量以及囊肿和纤维化体积下降。 Tolvaptan 显着提高心力衰竭大鼠的无电解质水清除率 (E-CH(2)O) 或水渗出至正值,并增加尿精氨酸加压素 (AVP) 排泄。
|
| 酶活实验 |
在体外受体结合研究中,托伐普坦阻断[(3)H]AVP与人V(2)受体的结合,其选择性是V(1a)受体的29倍,并且对V(1b)受体没有抑制作用。Tolvaptan不仅抑制[(3)H]AVP的结合,而且抑制AVP诱导的表达人V(2)受体的HeLa细胞中环状AMP的产生。此外,托伐普坦没有内在的V(2)受体激动作用[1]。
|
| 细胞实验 |
细胞系:HepG2 细胞
浓度:0-100 μM 孵育时间:24、48、96 和 168 小时 结果:对 HepG2 细胞具有时间和剂量依赖性抑制作用,IC50 >100、52.2、 24、48、96 和 168 小时分别为 33.0 和 27.1 μM。 |
| 动物实验 |
Male albino rats with cyclophosphamide intraperitoneal injection
10 mg/kg Oral gavage; 10 mg/kg once per day; for 22 days |
| 药代性质 (ADME/PK) |
Absorption, Distribution and Excretion
Tmax, Healthy subjects: 2 - 4 hours; Cmax, Healthy subjects, 30 mg: 374 ng/mL; Cmax, Healthy subjects, 90 mg: 418 ng/mL; Cmax, heart failure patients, 30 mg: 460 ng/mL; Cmax, heart failure patients, 90 mg: 723 ng/mL; AUC(0-24 hours), 60 mg: 3.71 μg·h/mL; AUC(∞), 60 mg: 4.55 μg·h/mL; The pharmacokinetic properties of tolvaptan are stereospecific, with a steady-state ratio of the S-(-) to the R-(+) enantiomer of about 3. The absolute bioavailability of tolvaptan is unknown. At least 40% of the dose is absorbed as tolvaptan or metabolites. Food does not impact the bioavailability of tolvaptan. Fecal- very little renal elimination (<1% is excreted unchanged in the urine) Healthy subjects: 3L/kg; slightly higher in heart failure patients. 4 mL/min/kg (post-oral dosing). In a study in patients with creatinine clearances ranging from 10-124 mL/min administered a single dose of 60 mg tolvaptan, AUC and Cmax of plasma tolvaptan were less than doubled in patients with severe renal impairment relative to the controls. The peak increase in serum sodium was 5-6 mEq/L, regardless of renal function, but the onset and offset of tolvaptan's effect on serum sodium were slower in patients with severe renal impairment. In healthy subjects the pharmacokinetics of tolvaptan after single doses of up to 480 mg and multiple doses up to 300 mg once daily have been examined. Area under the curve (AUC) increases proportionally with dose. After administration of doses > or = 60 mg, however, Cmax increases less than proportionally with dose. The pharmacokinetic properties of tolvaptan are stereospecific, with a steady-state ratio of the S-(-) to the R-(+) enantiomer of about 3. The absolute bioavailability of tolvaptan is unknown. At least 40% of the dose is absorbed as tolvaptan or metabolites. Peak concentrations of tolvaptan are observed between 2 and 4 hours post-dose. Food does not impact the bioavailability of tolvaptan. In vitro data indicate that tolvaptan is a substrate and inhibitor of P-gp. Tolvaptan is highly plasma protein bound (99%) and distributed into an apparent volume of distribution of about 3 L/kg. Tolvaptan is eliminated entirely by non-renal routes and mainly, if not exclusively, metabolized by CYP 3A. After oral dosing, clearance is about 4 mL/min/kg and the terminal phase half-life is about 12 hours. The accumulation factor of tolvaptan with the once-daily regimen is 1.3 and the trough concentrations amount to > or = 16% of the peak concentrations, suggesting a dominant half-life somewhat shorter than 12 hours. There is marked inter-subject variation in peak and average exposure to tolvaptan with a percent coefficient of variation ranging between 30 and 60%. In patients with hyponatremia of any origin the clearance of tolvaptan is reduced to about 2 mL/min/kg. Moderate or severe hepatic impairment or congestive heart failure decrease the clearance and increase the volume of distribution of tolvaptan, but the respective changes are not clinically relevant. Exposure and response to tolvaptan in subjects with creatinine clearance ranging between 79 and 10 mL/min and patients with normal renal function are not different. In healthy subjects receiving a single dose of Samsca 60 mg, the onset of the aquaretic and sodium increasing effects occurs within 2 to 4 hours post-dose. A peak effect of about a 6 mEq increase in serum sodium and about 9 mL/min increase in urine excretion rate is observed between 4 and 8 hours post-dose; thus, the pharmacological activity lags behind the plasma concentrations of tolvaptan. About 60% of the peak effect on serum sodium is sustained at 24 hours post-dose, but the urinary excretion rate is no longer elevated by this time. Doses above 60 mg tolvaptan do not increase aquaresis or serum sodium further. The effects of tolvaptan in the recommended dose range of 15 to 60 mg once daily appear to be limited to aquaresis and the resulting increase in sodium concentration. For more Absorption, Distribution and Excretion (Complete) data for Tolvaptan (12 total), please visit the HSDB record page. Metabolism / Metabolites Metabolism exclusively by CYP3A4 enzyme in the liver. Metabolites are inactive. Repeated dosing of female rats reduced systemic exposure to tolvaptan. Analysis of the serum samples for metabolites DM-4103 and DM-4107 revealed increases in the concentrations of these metabolites following repeated dosing, and explained the reduction in serum tolvaptan concentrations. Furthermore, tolvaptan was shown to induce hepatic drug-metabolising enzymes (cytochrome b5 content and aminopyrine N-demethylase activity) in female rats after 7 days dosing at 300 mg/kg/day. Tolvaptan was both a substrate for, and inhibitor of, MDR1-mediated transport. Tolvaptan is extensively metabolized in all species investigated. In vitro studies with rat liver supernatant produced a number of metabolites of tolvaptan. Hydroxylation of the benzazepine ring produced metabolites DM-4110, DM-4111 and DM-4119. Cleavage of the bond between the 1 and 2 positions of the benzazepine ring produced metabolites DM-4103, DM-4104, DM-4105 and DM- 4107. Oxidation of the hydroxyl group at the 5 position in the benzazepine ring produced MOP-21826. Tolvaptan is mainly, if not exclusively, metabolized in the liver by cytochrome P-450 (CYP) isoenzyme 3A; the drug also is a weak inhibitor of CYP3A and a substrate and inhibitor of the P-glycoprotein transport system. Compared with tolvaptan, metabolites of the drug have little or no antagonist activity for human V2 receptors. Tolvaptan is metabolized extensively in humans by the CYP3A4/5 system with seven metabolites (DM-4103, DM-4104, DM-4105, DM-4107, DM-4110, DM-4111, DM-4119) detected in the plasma, urine, and faeces of all subjects in a 14C mass balance study. After administration of (14)C-tolvaptan, 13 metabolites were identified in human plasma. Tolvaptan and identified metabolites accounted for about 70% of administered radioactivity. The predominant metabolite, with >50% of the total dose using the mass balance approach was DM-4103. The terminal elimination half-life of DM-4103 is approximatley 183 hours and after multiple dosing DM-4103 shows accumulation by day 28, but this appears pharmacologically inactive in the concentrations achieved using clinically relevant doses. Only 3% of the radioactivity was due to unchanged tolvaptan in the plasma. Biological Half-Life Terminal half life, oral dose = 12 hours. After IV administration, a half-life was estimated to be 3.5 hours, but it is suggested that this value most likely represents a distribution half-life and not a true elimination half-life. The terminal phase half-life is about 12 hours. Although tolvaptan is poorly soluble in water, following single dose of 30-480 mg, it is absorbed rapidly with a median time to peak plasma concentrations of about 2 hours (range of 1-12 hours) in healthy subjects. The mean (SD) of elimination half life is 7.8 (4.9) hours. |
| 毒性/毒理 (Toxicokinetics/TK) |
Toxicity Summary
IDENTIFICATION AND USE: Tolvaptan is a white crystalline powder that is formulated into oral tablets. Tolvaptan is an antagonist of arginine vasopressin (antidiuretic hormone) V2 receptors. It is used to treat low sodium levels in the blood. HUMAN EXPOSURE AND TOXICITY: Tolvaptan was well tolerated in healthy subjects at single oral doses up to 480 mg and multiple doses up to 300 mg once daily for 5 days. There is no specific antidote for tolvaptan intoxication. The signs and symptoms of an acute overdose can be anticipated to be those of excessive pharmacologic effect: a rise in serum sodium concentration, polyuria, thirst, and dehydration/hypovolemia. However, chronic administration of tolvaptan can cause serious and potentially fatal liver injury. In 2013, the U.S. Food and Drug Administration (FDA) determined that the drug should not be used for longer than 30 days and should not be used in patients with underlying liver disease because it can cause liver injury, potentially requiring liver transplant or death. In a placebo-controlled and open label extension study of chronically administered tolvaptan in patients with autosomal dominant polycystic kidney disease, cases of serious liver injury attributed to tolvaptan were observed. Tolvaptan therapy should be initiated or reinitiated only in a hospital setting, where serum sodium concentrations and therapeutic response can be monitored closely. Too rapid correction of hyponatremia may cause osmotic demyelination syndrome, resulting in dysarthria, mutism, dysphagia, lethargy, affective changes, spastic quadriparesis, seizures, coma, or death. Slower rates of correction may be advisable in susceptible patients, including those with severe malnutrition, alcoholism, or advanced liver disease. Patients with syndrome of inappropriate secretion of antidiuretic hormone or very low baseline serum sodium concentrations may be at increased risk for too rapid correction of serum sodium concentration. Tolvaptan is contraindicated in patients who are unable to sense or appropriately respond to thirst and in those with hypovolemic hyponatremia. Tolvaptan is mainly, if not exclusively, metabolized in the liver by cytochrome P-450 (CYP) isoenzyme 3A; the drug also is a weak inhibitor of CYP3A and a substrate and inhibitor of the P-glycoprotein transport system. Compared with tolvaptan, metabolites of the drug have little or no antagonist activity for human V2 receptors. ANIMAL STUDIES: Tolvaptan had low acute toxicity when administered to rats and dogs. In repeated dose studies in rats and dogs, findings were generally related to the pharmacological effect of tolvaptan and consisted of increased urine volume, decreased urine osmolality and increased water consumption. Decreased body weight and alterations in hematological and clinical chemistry parameters were also seen but were reversible during a recovery period. Up to two years of oral administration of tolvaptan to male and female rats did not increase the incidence of tumors. In a fertility study in male and female rats, tolvaptan was associated with fewer corpora lutea and implants compared to controls. Oral administration of tolvaptan to pregnant rabbits during organogenesis was associated with reductions in maternal body weight gain and food consumption. Abortions, increased incidences of embryo-fetal death, fetal microphthalmia, open eyelids, cleft palate, brachymelia, and skeletal malformations were also observed. Tolvaptan tested negative for genotoxicity in in vitro (bacterial reverse mutation assay and chromosomal aberration test in Chinese hamster lung fibroblast cells) and in vivo (rat micronucleus assay) test systems. Hepatotoxicity In prelicensure clinical trials, tolvaptan was not implicated in causing serum enzyme elevations or clinically apparent liver injury. However, instances of worsening of hepatic failure and complications of portal hypertension were reported in a small proportion of patients with cirrhosis treated with tolvaptan. These complications included variceal hemorrhage, hepatic encephalopathy and worsening of jaundice. In many trials, however, the frequency of these complications was not significantly greater than in placebo treated controls. More recently, in large registration trials of long term therapy in patients with ADPKD, serum aminotransferase elevations occurred in 4% to 5% of patients on tolvaptan, compared to only 1% of controls. Furthermore, clinically apparent liver injury occurred in approximately 0.1% of treated patients. The time to onset of illness ranged from 3 to 9 months (Case 1), but occasionally arose during long term therapy (Case 2). The clinical presentation was with the insidious development of fatigue, nausea and abdominal pain followed by dark urine, jaundice and pruritus. The pattern of serum enzyme elevations was typically hepatocellular or mixed, and liver biopsy showed an acute hepatitis with mild cholestasis. All patients recovered after stopping therapy, generally within 1 to 3 months of stopping therapy without evidence of residual injury. Immunoallergic features and autoantibodies were not found. Rapid recurrence on rechallenge was demonstrated in several patients with marked serum enzyme elevations during therapy, but patients with jaundice were not reexposed. The frequency of clinically apparent liver injury during therapy was one reason for the delay of formal approval of long term tolvaptan therapy for ADPKD. Since its approval and more wide-spread use, occasion reports of clinically apparent liver injury have continued to appear, at least one of which led to liver transplantation. Interestingly, most instances of liver injury have been reported with its use in autosomal dominant polycystic kidney disease rather than hyponatremia. Reasons for this are probably the duration of therapy, but also may relate to the slightly higher doses used to decrease progress in polycystic kidney disease. Likelihood score: C (probable rare cause of clinically apparent liver injury). Protein Binding 99% bound Interactions Tolvaptan, an arginine vasopressin (V2) receptor antagonist, may interfere with the V2 receptor agonist activity of desmopressin. In a male patient with mild von Willebrand disease, IV infusion of desmopressin 2 hours after oral administration of tolvaptan did not result in the expected increases in von Willebrand factor antigen or factor VIII activity. When the patient received desmopressin prior to initiation of tolvaptan therapy, desmopressin resulted in twofold to threefold increases in von Willebrand factor antigen, ristocetin cofactor activity, and factor VIII activity, and normalized results of the platelet function analyzer test and the activated partial thromboplastin time (aPTT). However, when the patient received desmopressin during tolvaptan therapy, desmopressin failed to increase von Willebrand factor antigen, ristocetin cofactor activity, and factor VIII activity, and effects of desmopressin on platelet function analyzer test results and aPTT were attenuated. Concomitant use of tolvaptan with V2 receptor agonists is not recommended. In clinical studies, the incidence of hyperkalemia was approximately 1-2% higher when tolvaptan was used concomitantly with angiotensin II receptor antagonists, angiotensin-converting enzyme (ACE) inhibitors, and potassium-sparing diuretics compared with use of these drugs with placebo. Formal drug interaction studies have not been performed. Serum potassium concentrations should be monitored during concomitant therapy with tolvaptan and drugs known to increase serum potassium concentrations (e.g., angiotensin II receptor antagonists, ACE inhibitors, potassium-sparing diuretics). Concomitant use of tolvaptan with inhibitors of the P-glycoprotein transport system (e.g., cyclosporine) may result in increased tolvaptan concentrations and may require reduction of tolvaptan dosage based on clinical response. Concomitant use of tolvaptan with potent inducers of CYP3A (e.g., barbiturates, carbamazepine, phenytoin, rifabutin, rifampin, rifapentine, St. John's wort (Hypericum perforatum)) may result in reduced plasma concentrations and decreased efficacy of tolvaptan. The manufacturer states that concomitant use of tolvaptan with rifampin reduces plasma tolvaptan concentrations by 85%, and other potent CYP3A inducers can be expected to produce similar results. Concomitant use of tolvaptan with CYP3A inducers should be avoided. If tolvaptan is used concomitantly with CYP3A inducers, the expected clinical effects of tolvaptan may not be observed at the recommended dosage; patient response should be monitored and the dosage adjusted accordingly. For more Interactions (Complete) data for Tolvaptan (8 total), please visit the HSDB record page. |
| 参考文献 | |
| 其他信息 |
Therapeutic Uses
Vasopressin V2 Receptor Antagonist Samsca is indicated for the treatment of clinically significant hypervolemic and euvolemic hyponatremia (serum sodium <125 mEq/L or less marked hyponatremia that is symptomatic and has resisted correction with fluid restriction), including patients with heart failure and Syndrome of Inappropriate Antidiuretic Hormone (SIADH). /Included in US product label/ EXPL Autosomal dominant polycystic kidney disease (ADPKD) is characterized by bilateral renal cysts, kidney pain, hypertension, and progressive loss of renal function. It is a leading cause of end-stage renal disease and the most common inherited kidney disease in the United States. Despite its prevalence, disease-modifying treatment options do not currently exist. Tolvaptan is an orally active, selective arginine vasopressin V2 receptor antagonist already in use for hyponatremia. Tolvaptan exhibits dose-proportional pharmacokinetics with a half-life of approximately 12 hours. Metabolism occurs through the cytochrome P450 3A4 isoenzyme, and tolvaptan is a substrate for P-glycoprotein, resulting in numerous drug interactions. Recent research has highlighted the beneficial effect of tolvaptan on delaying the progression of ADPKD, which is the focus of this review. Pharmacologic, preclinical, and phase II and III clinical trial studies have demonstrated that tolvaptan is an effective treatment option that targets underlying pathogenic mechanisms of ADPKD. Tolvaptan delays the increase in total kidney volume (surrogate marker for disease progression), slows the decline in renal function, and reduces kidney pain. However, tolvaptan has significant adverse effects including aquaretic effects (polyuria, nocturia, polydipsia) and elevation of aminotransferase enzyme concentrations with the potential for acute liver failure. Appropriate patient selection is critical to optimize long-term benefits while minimizing adverse effects and hepatotoxic risk factors. Overall, tolvaptan is the first pharmacotherapeutic intervention to demonstrate significant benefit in the treatment of ADPKD, but practitioners and regulatory agencies must carefully weigh the risks versus benefits. Additional research should focus on incidence and risk factors of liver injury, cost-effectiveness, clinical management of drug-drug interactions, and long-term disease outcomes. Tolvaptan is not indicated for the treatment of hypovolemic hyponatremia. The manufacturer states that tolvaptan should not be used in patients who require urgent intervention to raise serum sodium concentrations to prevent or treat serious neurologic manifestations. In addition, it has not been established that using tolvaptan to increase serum sodium concentrations provides symptomatic benefit to patients Drug Warnings /BOXED WARNING/ WARNING: INITIATE AND RE-INITIATE IN A HOSPITAL AND MONITOR SERUM SODIUM. Samsca should be initiated and re-initiated in patients only in a hospital where serum sodium can be monitored closely. Too rapid correction of hyponatremia (e.g., >12 mEq/L/24 hours) can cause osmotic demyelination resulting in dysarthria, mutism, dysphagia, lethargy, affective changes, spastic quadriparesis, seizures, coma and death. In susceptible patients, including those with severe malnutrition, alcoholism or advanced liver disease, slower rates of correction may be advisable. Tolvaptan therapy should be initiated or reinitiated only in a hospital setting, where serum sodium concentrations and therapeutic response can be monitored closely. Too rapid a correction of hyponatremia (e.g., increases in serum sodium concentration exceeding 12 mEq/L over 24 hours) may cause osmotic demyelination syndrome, resulting in dysarthria, mutism, dysphagia, lethargy, affective changes, spastic quadriparesis, seizures, coma, or death. Slower rates of correction may be advisable in susceptible patients, including those with severe malnutrition, alcoholism, or advanced liver disease. Patients with syndrome of inappropriate secretion of antidiuretic hormone (SIADH) or very low baseline serum sodium concentrations may be at increased risk for too rapid a correction of serum sodium concentration. Fluid restriction during the first 24 hours of tolvaptan therapy may increase the risk of overly rapid correction of serum sodium concentration and generally should be avoided. Samsca can cause serious and potentially fatal liver injury. In a placebo-controlled and open label extension study of chronically administered tolvaptan in patients with autosomal dominant polycystic kidney disease, cases of serious liver injury attributed to tolvaptan were observed. An increased incidence of ALT greater than three times the upper limit of normal was associated with tolvaptan (42/958 or 4.4%) compared to placebo (5/484 or 1.0%). Cases of serious liver injury were generally observed starting 3 months after initiation of tolvaptan although elevations of ALT occurred prior to 3 months. Patients with symptoms that may indicate liver injury, including fatigue, anorexia, right upper abdominal discomfort, dark urine or jaundice should discontinue treatment with Samsca. Limit duration of therapy with Samsca to 30 days. Avoid use in patients with underlying liver disease, including cirrhosis, because the ability to recover from liver injury may be impaired The U.S. Food and Drug Administration (FDA) has determined that the drug Samsca (tolvaptan) should not be used for longer than 30 days and should not be used in patients with underlying liver disease because it can cause liver injury, potentially requiring liver transplant or death. Samsca is used to treat low sodium levels in the blood. An increased risk of liver injury was observed in recent large clinical trials evaluating Samsca for a new use in patients with autosomal dominant polycystic kidney disease (ADPKD) For more Drug Warnings (Complete) data for Tolvaptan (15 total), please visit the HSDB record page. Pharmacodynamics Urine volume and fluid intake increase in a dose dependent manner which results in overall negative fluid balance in patients taking tolvaptan. Increases in serum sodium and osmolality can be observed 4-8 hours post-administration and is maintained for 24 hours. The magnitude of serum sodium and osmolality change increases with escalating doses. Furthermore, a decrease in urine osmolality and increase in free water clearance can be observed 4 hours after post-administration of tolvaptan. The affinity for V2 receptors is 29x greater than that of V1a receptors and does not have any appreciable affinity for V2 receptors. |
| 分子式 |
C26H25CLN2O3
|
|---|---|
| 分子量 |
448.94
|
| 精确质量 |
448.155
|
| 元素分析 |
C, 69.56; H, 5.61; Cl, 7.90; N, 6.24; O, 10.69
|
| CAS号 |
150683-30-0
|
| 相关CAS号 |
Tolvaptan-d7; 1246818-18-7
|
| PubChem CID |
216237
|
| 外观&性状 |
White to off-white solid powder
|
| 密度 |
1.3±0.1 g/cm3
|
| 沸点 |
594.4±50.0 °C at 760 mmHg
|
| 熔点 |
219-222°C
|
| 闪点 |
313.3±30.1 °C
|
| 蒸汽压 |
0.0±1.8 mmHg at 25°C
|
| 折射率 |
1.664
|
| LogP |
4.09
|
| tPSA |
69.64
|
| 氢键供体(HBD)数目 |
2
|
| 氢键受体(HBA)数目 |
3
|
| 可旋转键数目(RBC) |
3
|
| 重原子数目 |
32
|
| 分子复杂度/Complexity |
674
|
| 定义原子立体中心数目 |
0
|
| SMILES |
ClC1C([H])=C([H])C2=C(C=1[H])C([H])(C([H])([H])C([H])([H])C([H])([H])N2C(C1C([H])=C([H])C(=C([H])C=1C([H])([H])[H])N([H])C(C1=C([H])C([H])=C([H])C([H])=C1C([H])([H])[H])=O)=O)O[H]
|
| InChi Key |
GYHCTFXIZSNGJT-UHFFFAOYSA-N
|
| InChi Code |
InChI=1S/C26H25ClN2O3/c1-16-6-3-4-7-20(16)25(31)28-19-10-11-21(17(2)14-19)26(32)29-13-5-8-24(30)22-15-18(27)9-12-23(22)29/h3-4,6-7,9-12,14-15,24,30H,5,8,13H2,1-2H3,(H,28,31)
|
| 化学名 |
N-[4-(7-chloro-5-hydroxy-2,3,4,5-tetrahydro-1-benzazepine-1-carbonyl)-3-methylphenyl]-2-methylbenzamide
|
| 别名 |
OPC41061; Tolvaptan; OPC 41061; OPC-41061; trade names Samsca; Jinarc; Resodim
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
|
|||
|---|---|---|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 2.17 mg/mL (4.83 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 21.7 mg/mL澄清DMSO储备液加入400 μL PEG300中,混匀;然后向上述溶液中加入50 μL Tween-80,混匀;加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: ≥ 2.17 mg/mL (4.83 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 21.7 mg/mL澄清DMSO储备液加入900 μL玉米油中,混合均匀。 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.2275 mL | 11.1373 mL | 22.2747 mL | |
| 5 mM | 0.4455 mL | 2.2275 mL | 4.4549 mL | |
| 10 mM | 0.2227 mL | 1.1137 mL | 2.2275 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
| NCT Number | Recruitment | interventions | Conditions | Sponsor/Collaborators | Start Date | Phases |
| NCT06171100 | Recruiting | Drug: Tolvaptan | Hyponatremia SIADH |
King's College Hospital NHS Trust | December 15, 2023 | N/A |
| NCT05569655 | Recruiting | Drug: Tolvaptan Drug: Furosemide |
Pulmonary Arterial Hypertension Randomized Controlled Trial |
Chinese Pulmonary Vascular Disease Research Group |
April 6, 2021 | Not Applicable |
| NCT04782258 | Recruiting | Drug: Tolvaptan Suspension Drug: Tolvaptan Tablets |
ARPKD | Otsuka Pharmaceutical Development & Commercialization, Inc. |
July 15, 2022 | Phase 3 |
| NCT03406286 | Recruiting | Drug: Tolvaptan | Safety | Korea Otsuka Pharmaceutical Co., Ltd. |
July 19, 2016 | N/A |
| NCT04790175 | Recruiting | Drug: Tolvaptan (SAMSCA) |
Antidiuretic Hormone, Inappropriate Secretion |
Otsuka Pharmaceutical Co., Ltd. | March 29, 2021 | N/A |
|
|
|
|
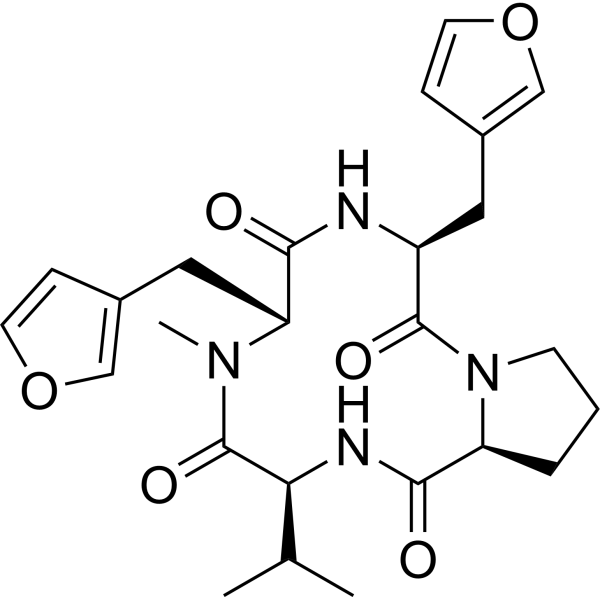 Endolide F
Endolide F
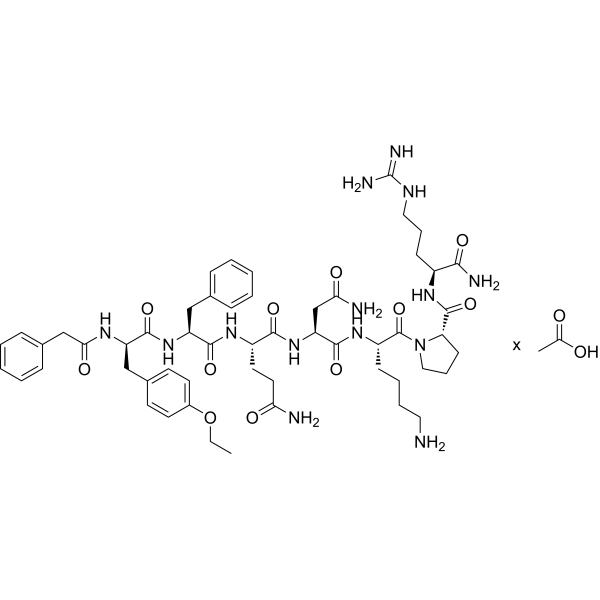 (Phenylac1,D-Tyr(Et)2,Lys6,Arg8,des-Gly9)-Vasopressin acetate
(Phenylac1,D-Tyr(Et)2,Lys6,Arg8,des-Gly9)-Vasopressin acetate
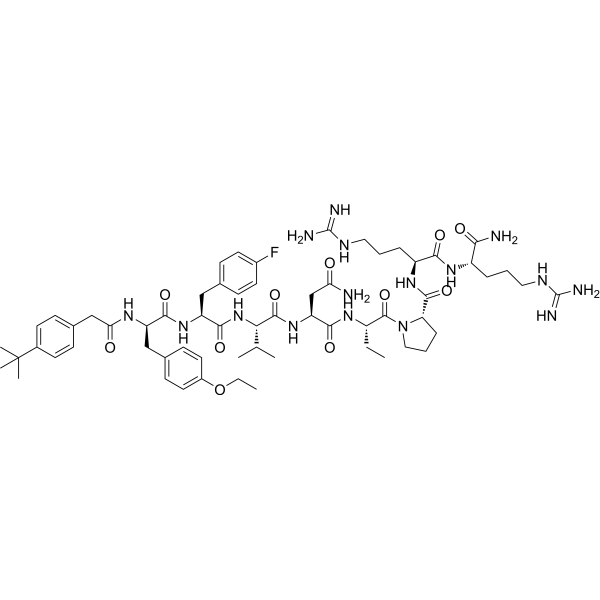 Vasopressin V2 receptor antagonist 2
Vasopressin V2 receptor antagonist 2
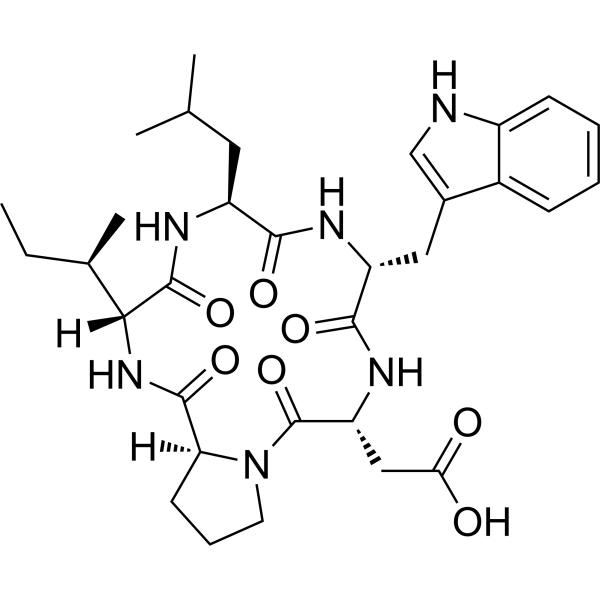 JKC 301
JKC 301
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1






























 COA
COA




