| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 10 mM * 1 mL in DMSO |
|
||
| 1mg |
|
||
| 5mg |
|
||
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| 500mg |
|
||
| 1g |
|
||
| Other Sizes |
|

| 靶点 |
JAK3 (IC50 = 1 nM); JAK2 (IC50 = 20 nM); JAK1 (IC50 = 112 nM); Rock-II (IC50 = 3400 nM); Lck (IC50 = 3870 nM)
|
||
|---|---|---|---|
| 体外研究 (In Vitro) |
JAK3 和 JAK2 可以与浓度为 2.2 nM 和 5 nM (Kd) 的托法替布 (CP-690550) 柠檬酸盐结合。托法替尼包含在 Camk1 (Kd 5,000 nM)、DCamkL3 (Kd 4.5 nM)、Mst2 (Kd 4,300 nM)、Pkn1 (Kd 200 nM) 和 Rps6ka2 (Kin.Dom.2-C-) 中。终端的 Kd 1,400 nM)、Rps6ka6(Kin.Dom.2-C 终端)的 Kd 1,200 nM、Snark 的 Kd 420 nM、Tnk1 的 Kd 640 nM 和 Tyk2][1] 的 Kd 620 nM。
|
||
| 体内研究 (In Vivo) |
初次免疫后五周(p<0.01,n=8),与 PEG 治疗的对照小鼠相比,用tofacitinib/托法替尼治疗的动物表现出抗药物抗体 (ADA) 的发育显着下降。此外,第 28 天是 ADA 变得引人注目的时候。 SS1P 滴度从第 21 天到第 35 天分别显示出 1000 到 200 倍的变化。注射匙孔血蓝蛋白 (KLH) 的动物比接受 SS1P 治疗的动物更快地产生抗体反应。然而,与对照组相比,tofacitinib/托法替尼剂量降低了抗 KLH 滴度(分别在第 21 天 p<0.05 和第 28 天 p<0.01,n = 5)。从第 21 天到第 28 天,滴度降低了 5000 到 250 倍[2]。每日剂量为 6.2 mg/kg 的tofacitinib/托法替尼可以抑制 JAK1 和 JAK3 信号通路 4 小时以上,该剂量是根据之前的剂量反应实验选择的,对后爪体积和血浆暴露有 80% 的抑制作用[3 ]。
口服tofacitinib/托法替尼可抑制LPS诱导的气道中性粒细胞增多、BALF中某些细胞因子的水平以及肺组织中STAT3的磷酸化。 结论和意义:总之,本研究表明JAK抑制改善了LPS诱导的大鼠吸入性气道炎症,表明JAK/STAT3信号至少参与了LPS诱导肺中性粒细胞增多症的建立。应进一步研究JAKs抑制剂作为呼吸道炎症性疾病的潜在治疗方法。[4] 免疫原性仍然是基于蛋白质的疗法的“阿喀琉斯之踵”。针对蛋白质治疗产生的抗药物抗体会严重限制这类不断扩大的药物的安全性和有效性。在这篇文章中,我们报告了用tofacitinib/托法替尼(JAK抑制剂)对小鼠进行单一治疗,可以抑制对细菌蛋白假单胞菌外毒素A衍生的免疫毒素以及对银钥匙孔血蓝蛋白模型的抗体反应。免疫后21天,观察到两种Ag的IgG1滴度降低了数千倍。事实上,所有IgG同种型和IgM都明显受到抑制。胸腺非依赖性II型抗原的IgG3产生也减少。机制研究表明,tofacitinib/托法替尼治疗导致CD127+pro-B细胞数量减少。此外,我们观察到用tofacitinib/托法替尼治疗的小鼠生发中心B细胞减少,生发中心形成受损。由于托法替尼治疗期间仍存在正常的Ig水平,因此该药物特异性降低了抗药物Abs,从而保留了生物疗法的潜在疗效,包括用作癌症疗法的疗效[2]。 |
||
| 酶活实验 |
激酶谱由CRO公司利用KINOMEscan™进行。通过与专有标签融合的选定激酶的竞争结合分析记录活性。在存在和不存在测试化合物的情况下,对结合到固定的活性位点导向配体的激酶量的测量提供了配体结合的DMSO对照百分比。选择0至10之间的活性进行Kd测定。树状图表示是由名为PhyloChem的内部可视化工具生成的。树状图聚类和顶点基于人类系统发育激酶数据,可在http://kinase.com/human/kinome1.
|
||
| 细胞实验 |
B细胞分化和增殖的特征[2]
从BALB/c小鼠制备脾细胞和骨髓细胞悬浮液,并计数总细胞。细胞(1×106)用以下抗小鼠抗体的各种组合染色:CD3、B220、CD43、IgM、Fas、GL-7、CD24、BP-1、CD127或IgG1与FITC、PE或APC结合,并在FACSCalibur流式细胞仪上分析。至少获得了10000个现场活动。为了评估体外B细胞增殖,根据制造商的说明,将CD43-脾细胞纯化为MACS,并用1μM CFSE标记。在0、0.1、0.3或1.0μM的托法替尼存在下,用25μg/mL的LPS(大肠杆菌0111:B4)和5ng/mL的IL-4激活标记细胞48小时。培养后,洗涤细胞,表面染色,并用流式细胞术检查。 体外人破骨细胞分化和功能。[3] 通过磁激活细胞分选(MACS)细胞分离技术从白细胞包中阴性选择CD14+细胞,获得原代人单核细胞。将细胞以1×105个细胞/孔的速度接种在96孔黑色组织培养板上,在含有5%胎牛血清(FBS)和10单位/ml青霉素-链霉素的高糖Dulbecco改良Eagle培养基中。每隔一天用25ng/ml重组人巨噬细胞集落刺激因子(M-CSF)处理培养的细胞14天,用于巨噬细胞分化,或在100ng/ml重组人RANKL存在下用M-CSF处理破骨细胞分化。在分化细胞因子的同时,还用或不用0.2%DMSO中不同浓度的托法替尼处理细胞。使用ELF-97荧光磷酸酶底物定量TRAP活性。然后根据制造商的建议,使用白细胞酸性磷酸酶试剂盒固定细胞并染色。[3] 通过溶骨试验测定人破骨细胞的功能性骨吸收活性。将人破骨细胞前体细胞 以1×104个细胞/孔的速度接种在含有33 ng/ml M-CSF和66 ng/ml RANKL的培养基中,然后接种在预涂有铕结合的人I型胶原的96孔OsteoLyse细胞培养板上。在分化阶段(第0-6天),细胞用不同浓度的托法替尼处理或不处理。培养6天后,加入含有M-CSF和RANKL的新鲜培养基,并以相同浓度替换或加入先前未处理的细胞中。此外,将阿仑膦酸钠添加到未处理的细胞中作为阳性对照。将细胞再培养4天,以允许功能活性破骨细胞释放胶原蛋白,并使用OsteoLyse荧光细胞释放试剂检测培养上清液中的铕荧光,在340 nm激发和615 nm发射下测量400μs间隔内的时间分辨荧光。 体外人T淋巴细胞RANKL的产生。[3] 使用MACS细胞分离技术从leukpak中阴性选择CD4+T淋巴细胞,并在含有葡萄糖、10%FBS和10单位/ml青霉素-链霉素的RPMI 1640培养基中,在圆底96孔组织培养板中以2.5×105个细胞/孔的速度培养。细胞在0.2%DMSO中用或不用不同浓度的托法替尼处理,并用1μg/ml抗人CD3和0.1μg/ml抗人类CD28抗体以及50ng/ml重组人IL-2活化5天。使用人LincoPlex测定法测量分泌到培养基中的RANKL。 |
||
| 动物实验 |
|
||
| 药代性质 (ADME/PK) |
Absorption, Distribution and Excretion
74% oral absorption (absolute bioavailability), with peak plasma concentrations (T max) achieved in 0.5-1 hour. Administration with fatty meals does not alter AUC but reduces Cmax by 32%. 70% metabolized in the liver by CYP3A4 (major) and CYP2C19 (minor). Metabolites produced are inactive. 30% renally eliminated as unchanged drug. Vd= 87L after intravenous administration. Distribution is equal between red blood cells and plasma. The protein binding of tofacitinib is approximately 40%. Tofacitinib binds predominantly to albumin and does not appear to bind to a1-acid glycoprotein. Tofacitinib distributes equally between red blood cells and plasma. The absolute oral bioavailability of tofacitinib is 74%. Coadministration of Xeljanz with a high-fat meal resulted in no changes in AUC while Cmax was reduced by 32%. In clinical trials, Xeljanz was administered without regard to meals. Clearance mechanisms for tofacitinib are approximately 70% hepatic metabolism and 30% renal excretion of the parent drug. The metabolism of tofacitinib is primarily mediated by CYP3A4 with minor contribution from CYP2C19. In a human radiolabeled study, more than 65% of the total circulating radioactivity was accounted for by unchanged tofacitinib, with the remaining 35% attributed to 8 metabolites, each accounting for less than 8% of total radioactivity. The pharmacologic activity of tofacitinib is attributed to the parent molecule. Following oral administration of Xeljanz /to humans/, peak plasma concentrations are reached within 0.5-1 hour, elimination half-life is approximately 3 hours and a dose-proportional increase in systemic exposure was observed in the therapeutic dose range. Steady state concentrations are achieved in 24-48 hours with negligible accumulation after twice daily administration. /MILK/ Tofacitinib was secreted in milk of lactating rats. It is not known whether tofacitinib is excreted in human milk. Metabolism / Metabolites Metabolized in the liver by CYP3A4 and CYP2C19. Metabolites produced are inactive. Clearance mechanisms for tofacitinib are approximately 70% hepatic metabolism and 30% renal excretion of the parent drug. The metabolism of tofacitinib is primarily mediated by CYP3A4 with minor contribution from CYP2C19. In a human radiolabeled study, more than 65% of the total circulating radioactivity was accounted for by unchanged tofacitinib, with the remaining 35% attributed to 8 metabolites, each accounting for less than 8% of total radioactivity. The pharmacologic activity of tofacitinib is attributed to the parent molecule. Biological Half-Life ~3 hours The elimination half-life of tofacitinib /in humans/ is approximately 3 hours. |
||
| 毒性/毒理 (Toxicokinetics/TK) |
Toxicity Summary
IDENTIFICATION AND USE: Tofacitinib is a yellow foam. As the drug Xeljanz, it is indicated for the treatment of adult patients with moderately to severely active rheumatoid arthritis (RA) who have had an inadequate response or intolerance to methotrexate. It may be used as monotherapy or in combination with methotrexate or other nonbiologic disease-modifying antirheumatic drugs (DMARDs). HUMAN EXPOSURE AND TOXICITY: According to epidemiological studies, the overall risk of infection (including serious infection) and mortality rates in RA patients treated with tofacitinib appear to be similar to those observed in RA patients treated with biologic agents. The rates of serious infection were stable over time. Within the global tofacitinib RA development program, tuberculosis was the most common opportunistic infection reported but was rare in regions of low and medium TB incidence. In a genotoxicity study, increases in chromosomal abnormalities were observed in a human lymphocyte in vitro cytogenetic assay, at high cytotoxic concentrations with metabolic activation, but no effects were observed without metabolic activation. ANIMAL STUDIES: In cynomolgus monkeys, emesis and decreased activity were observed as a result of acute exposure. Xeljanz caused death in rats at single oral doses of >/= 500 mg/kg. Immune and hematopoietic organ systems were identified as main targets in repeat-dose toxicity studies on animals. In a peri/postnatal development study in rats, Xeljanz decreased the number of delivered and live born pups, and reduced pup survival at oral doses of 50 mg/kg/day. Xeljanz was teratogenic (external, visceral and skeletal abnormalities) in rabbits and rats at oral doses of 30 and 100 mg/kg/day, respectively. Xeljanz was not mutagenic in the bacterial reverse mutation assay. Xeljanz was not mutagenic in mammalian cells (in vitro CHO/HGPRT assay) and did not induce primary DNA damage in an in vivo/in vitro rat hepatocyte unscheduled DNA synthesis assay. Xeljanz was also negative in the in vivo rat micronucleus test. In a 2-year rat carcinogenicity study, Xeljanz induced benign Leydig cell tumors and malignant hibernomas at oral doses of >/= 30 mg/kg/day and benign thymomas at 100/75 mg/kg/day. In a 39-week repeat-dose toxicity study in adult monkeys, lymphomas were observed at the high dose of 5 mg/kg twice daily, but not at the lower dose of 1 mg/kg twice daily (approximately equivalent to human exposure). Interactions In healthy individuals, the CYP3A inducer rifampin (600 mg orally once daily for 7 days) decreased peak plasma concentrations and AUC of tofacitinib (single oral dose of 30 mg) by 74 and 84%, respectively. Concomitant use of rifampin may decrease efficacy of tofacitinib. There is a risk of added immunosuppression when Xeljanz is coadministered with potent immunosuppressive drugs (e.g., azathioprine, tacrolimus, cyclosporine). Combined use of multiple-dose Xeljanz with potent immunosuppressants has not been studied in rheumatoid arthritis. Use of Xeljanz in combination with biologic disease-modifying antirheumatic drugs (DMARDS) or potent immunosuppressants such as azathioprine and cyclosporine is not recommended. Concomitant use of tofacitinib with potent immunosuppressive agents (e.g., azathioprine, cyclosporine, tacrolimus) increases the risk of immunosuppression and is not recommended. Concomitant use of tofacitinib with such agents in patients with rheumatoid arthritis has not been studied to date. In healthy individuals, cyclosporine (200 mg orally every 12 hours for 5 days) decreased the clearance of tofacitinib (single oral dose of 10 mg), resulting in a 73% increase in the AUC of tofacitinib, accompanied by a 17% decrease in peak plasma tofacitinib concentrations. In healthy individuals, tacrolimus (5 mg orally every 12 hours for 7 days) slightly decreased the clearance of tofacitinib (single oral dose of 10 mg), resulting in a 21% increase in the AUC of tofacitinib, accompanied by a 9% decrease in peak plasma tofacitinib concentrations. Tofacitinib exposure is decreased when Xeljanz is coadministered with potent CYP3A4 inducers (e.g., rifampin). For more Interactions (Complete) data for Tofacitinib (9 total), please visit the HSDB record page. |
||
| 参考文献 |
|
||
| 其他信息 |
Therapeutic Uses
Protein Kinase Inhibitors /CLINICAL TRIALS/ ClinicalTrials.gov is a registry and results database of publicly and privately supported clinical studies of human participants conducted around the world. The Web site is maintained by the National Library of Medicine (NLM) and the National Institutes of Health (NIH). Each ClinicalTrials.gov record presents summary information about a study protocol and includes the following: Disease or condition; Intervention (for example, the medical product, behavior, or procedure being studied); Title, description, and design of the study; Requirements for participation (eligibility criteria); Locations where the study is being conducted; Contact information for the study locations; and Links to relevant information on other health Web sites, such as NLM's MedlinePlus for patient health information and PubMed for citations and abstracts for scholarly articles in the field of medicine. Tofacitinib is included in the database. Xeljanz (tofacitinib) is indicated for the treatment of adult patients with moderately to severely active rheumatoid arthritis who have had an inadequate response or intolerance to methotrexate. It may be used as monotherapy or in combination with methotrexate or other nonbiologic disease-modifying antirheumatic drugs (DMARDs). /Included in US product label/ EXPL Tofacitinib is an oral Janus kinase inhibitor that is being investigated for psoriasis and psoriatic arthritis. Japanese patients aged 20 years or more with moderate to severe plaque psoriasis and/or psoriatic arthritis were double-blindly randomized 1:1 to tofacitinib 5 or 10 mg b.i.d. for 16 weeks, open-label 10 mg b.i.d. for 4 weeks, then variable 5 or 10 mg b.i.d. to Week 52. Primary end-points at Week 16 were the proportion of patients achieving at least a 75% reduction in Psoriasis Area and Severity Index (PASI75) and Physician's Global Assessment of "clear" or "almost clear" (PGA response) for psoriasis, and 20% or more improvement in American College of Rheumatology criteria (ACR20) for patients with psoriatic arthritis. Safety was assessed throughout. Eighty-seven patients met eligibility criteria for moderate to severe plaque psoriasis (5 mg b.i.d., n = 43; 10 mg b.i.d., n = 44), 12 met eligibility criteria for psoriatic arthritis (5 mg b.i.d., n = 4; 10 mg b.i.d., n = 8) including five who met both criteria (10 mg b.i.d.). At Week 16, 62.8% and 72.7% of patients achieved PASI75 with tofacitinib 5 and 10 mg b.i.d., respectively; 67.4% and 68.2% achieved PGA responses; all patients with psoriatic arthritis achieved ACR20. Responses were maintained through Week 52. Adverse events occurred in 83% of patients through Week 52, including four (4.3%) serious adverse events and three (3.2%) serious infections (all herpes zoster). No malignancies, cardiovascular events or deaths occurred. Tofacitinib (both doses) demonstrated efficacy in patients with moderate to severe plaque psoriasis and/or psoriatic arthritis through 52 weeks; safety findings were generally consistent with prior studies. EXPL The inflammatory diseases ulcerative colitis and Crohn's disease constitute the two main forms of inflammatory bowel disease (IBD). They are characterized by chronic, relapsing inflammation of the gastrointestinal tract, significantly impacting on patient quality of life and often requiring prolonged treatment. Existing therapies for IBD are not effective for all patients, and an unmet need exists for additional therapies to induce and maintain remission. Here we describe the mechanism of action of the Janus kinase (JAK) inhibitor, tofacitinib, for the treatment of IBD and the effect of JAK inhibition on the chronic cycle of inflammation that is characteristic of the disease. The pathogenesis of IBD involves a dysfunctional response from the innate and adaptive immune system, resulting in overexpression of multiple inflammatory cytokines, many of which signal through JAKs. Thus JAK inhibition allows multiple cytokine signaling pathways to be targeted and is expected to modulate the innate and adaptive immune response in IBD, thereby interrupting the cycle of inflammation. Tofacitinib is an oral, small molecule JAK inhibitor that is being investigated as a targeted immunomodulator for IBD. Clinical development of tofacitinib and other JAK inhibitors is ongoing, with the aspiration of providing new treatment options for IBD that have the potential to deliver prolonged efficacy and clinically meaningful patient benefits. Drug Warnings /BOXED WARNING/ WARNING: SERIOUS INFECTIONS. Patients treated with Xeljanz are at increased risk for developing serious infections that may lead to hospitalization or death. Most patients who developed these infections were taking concomitant immunosuppressants such as methotrexate or corticosteroids. If a serious infection develops, interrupt Xeljanz until the infection is controlled. Reported infections include: Active tuberculosis, which may present with pulmonary or extrapulmonary disease. Patients should be tested for latent tuberculosis before Xeljanz use and during therapy. Treatment for latent infection should be initiated prior to Xeljanz use. Invasive fungal infections, including cryptococcosis and pneumocystosis. Patients with invasive fungal infections may present with disseminated, rather than localized, disease. Bacterial, viral, and other infections due to opportunistic pathogens. The risks and benefits of treatment with Xeljanz should be carefully considered prior to initiating therapy in patients with chronic or recurrent infection. Patients should be closely monitored for the development of signs and symptoms of infection during and after treatment with Xeljanz, including the possible development of tuberculosis in patients who tested negative for latent tuberculosis infection prior to initiating therapy /BOXED WARNING/ MALIGNANCIES. Lymphoma and other malignancies have been observed in patients treated with Xeljanz. Epstein Barr Virus-associated post-transplant lymphoproliferative disorder has been observed at an increased rate in renal transplant patients treated with Xeljanz and concomitant immunosuppressive medications Patients receiving tofacitinib are at increased risk of developing serious infections that may require hospitalization or result in death. Opportunistic infections caused by bacterial, mycobacterial, invasive fungal, viral, or other opportunistic organisms-including cryptococcosis, pneumocystosis, tuberculosis and other mycobacterial infections, esophageal candidiasis, multidermatomal herpes zoster, cytomegalovirus infection, and BK virus infection-have been reported in patients with rheumatoid arthritis receiving tofacitinib. Patients with invasive fungal infections may present with disseminated, rather than localized, disease. Patients should be closely monitored during and after treatment with tofacitinib for the development of signs or symptoms of infection (e.g., fever, malaise, weight loss, sweats, cough, dyspnea, pulmonary infiltrates, serious systemic illness including shock). Most patients who developed serious infections were receiving concomitant therapy with immunosuppressive agents such as methotrexate or corticosteroids. Tofacitinib therapy should not be initiated in patients with active infections, including localized infections. Tofacitinib should be discontinued in patients who develop a serious infection, opportunistic infection, or sepsis and should not be resumed until the infection is controlled. Clinicians should consider potential risks and benefits of tofacitinib prior to initiating therapy in patients with a history of chronic, recurring, serious, or opportunistic infections; patients with underlying conditions that may predispose them to infections; and patients who have been exposed to tuberculosis or who reside or have traveled in regions where tuberculosis or mycoses are endemic. Any patient who develops a new infection while receiving tofacitinib should undergo a thorough diagnostic evaluation (appropriate for an immunocompromised patient), appropriate anti-infective therapy should be initiated, and the patient should be closely monitored. For more Drug Warnings (Complete) data for Tofacitinib (22 total), please visit the HSDB record page. Pharmacodynamics Tofacitinib targets inflammation present in rheumatoid arthritis by inhibiting the janus kinases involved in the inflammatory response pathway. In placebo controlled trials of rheumatoid arthritis patients receiving 5mg or 10mg of tofacitinib twice daily, higher ACR20 responses were observed within 2 weeks in some patients (with ACR20 being defined as a minimum 20% reduction in joint pain or tenderness and 20% reduction in arthritis pain, patient disability, inflammatory markers, or global assessments of arthritis by patients or by doctors, according to the American College of Rheumatology (ACR) response criteria list), and improvements in physical functioning greater than placebo were also noted. Common known adverse effects of tofacitinib include headaches, diarrhea, nausea, nasopharyngitis and upper respiratory tract infection. More serious immunologic and hematological adverse effects have also been noted resulting in lymphopenia, neutropenia, anemia, and increased risk of cancer and infection. Before initiations of tofacitinib patients should be tested for latent infections of tuberculosis, and should be closely monitored for signs and symptoms of infection (fungal, viral, bacterial, or mycobacterial) during therapy. Therapy is not to be started in the presence of active infection, systemic or localized, and is to be interrupted if a serious infection occurs. Tofacitinib has been associated with an increased risk of lymphomas, such as Epstein-Barr virus associated lymphomas, and other malignancies (including lung, breast, gastric, and colorectal cancers). It is recommended to monitor lymphocytes, neutrophils, hemoglobin, liver enzymes, and lipids. Tofacitinib use is associated with a rapid decrease in C-reactive protein (CRP), dose dependent decreases in natural killer cells, and dose dependent increases in B cells. Depression in C-reactive protein levels continue after 2 weeks of tofacitinib discontinuation and suggest that pharmacodynamic activity last longer than pharmacokinetic half life. |
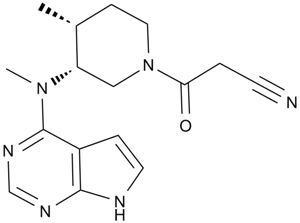
| 分子式 |
C16H20N6O
|
|---|---|
| 分子量 |
312.37
|
| 精确质量 |
312.169
|
| 元素分析 |
C, 61.52; H, 6.45; N, 26.90; O, 5.12
|
| CAS号 |
477600-75-2
|
| 相关CAS号 |
Tofacitinib citrate;540737-29-9;(3S,4S)-Tofacitinib;1092578-47-6;(3R,4S)-Tofacitinib;1092578-46-5;(3S,4R)-Tofacitinib;1092578-48-7;Tofacitinib-13C3; 1443435-54-8 (oxalate); 477600-75-2; 1803005-18-6 (HCl); 1443435-50-4 (tartrate); 2052885-67-1; 1803005-19-7 (HBr)
|
| PubChem CID |
9926791
|
| 外观&性状 |
Off-white to light yellow solid powder
|
| 密度 |
1.3±0.1 g/cm3
|
| 沸点 |
585.8±50.0 °C at 760 mmHg
|
| 熔点 |
White crystalline solid. MP: 199-206 °C /Tofacitinib monocitrate/
|
| 闪点 |
308.1±30.1 °C
|
| 蒸汽压 |
0.0±1.6 mmHg at 25°C
|
| 折射率 |
1.646
|
| LogP |
0.93
|
| tPSA |
88.91
|
| 氢键供体(HBD)数目 |
1
|
| 氢键受体(HBA)数目 |
5
|
| 可旋转键数目(RBC) |
3
|
| 重原子数目 |
23
|
| 分子复杂度/Complexity |
488
|
| 定义原子立体中心数目 |
2
|
| SMILES |
C[C@@H]1CCN(C[C@@H]1N(C)C2=NC=NC3=C2C=CN3)C(=O)CC#N
|
| InChi Key |
UJLAWZDWDVHWOW-YPMHNXCESA-N
|
| InChi Code |
InChI=1S/C16H20N6O/c1-11-5-8-22(14(23)3-6-17)9-13(11)21(2)16-12-4-7-18-15(12)19-10-20-16/h4,7,10-11,13H,3,5,8-9H2,1-2H3,(H,18,19,20)/t11-,13+/m1/s1
|
| 化学名 |
3-((3R,4R)-4-Methyl-3-(methyl(7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino)piperidin-1-yl)-3-oxopropanenitrile
|
| 别名 |
CP-690550; CP690550; CP 690550; Tasocitinib; Tasocitinib; 3-((3R,4R)-4-methyl-3-(methyl(7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino)piperidin-1-yl)-3-oxopropanenitrile; CP-690550; CP 690550; 1259404-17-5; rac-Tofacitinib; Tofacitinib; Xeljanz (Trade name); Tofacitinib free base;
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
|
|||
|---|---|---|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 2.5 mg/mL (8.00 mM) (饱和度未知) in 5% DMSO + 40% PEG300 + 5% Tween80 + 50% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
*生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: ≥ 2.5 mg/mL (8.00 mM) (饱和度未知) in 5% DMSO + 95% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 View More
配方 3 中的溶解度: ≥ 2.08 mg/mL (6.66 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 配方 4 中的溶解度: ≥ 2.08 mg/mL (6.66 mM) (饱和度未知) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将100μL 20.8mg/mL澄清的DMSO储备液加入到900μL 20%SBE-β-CD生理盐水中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 配方 5 中的溶解度: ≥ 2.08 mg/mL (6.66 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将100 μL 20.8 mg/mL 澄清 DMSO 储备液加入900 μL 玉米油中,混合均匀。 配方 6 中的溶解度: 30% PEG400+0.5% Tween80+5% propylene glycol:30mg/mL 配方 7 中的溶解度: 5 mg/mL (16.01 mM) in 0.5% MC 0.5% Tween-80 (这些助溶剂从左到右依次添加,逐一添加), 悬浊液; 超声助溶。 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 3.2013 mL | 16.0067 mL | 32.0133 mL | |
| 5 mM | 0.6403 mL | 3.2013 mL | 6.4027 mL | |
| 10 mM | 0.3201 mL | 1.6007 mL | 3.2013 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
| NCT Number | Recruitment | interventions | Conditions | Sponsor/Collaborators | Start Date | Phases |
| NCT06202560 | Enrolling by invitation | Drug: Tofacitinib 5 MG | Frontal Fibrosing Alopecia Lichen Planopilaris |
Institute of Dermatology, Thailand | November 29, 2023 | Not Applicable |
| NCT06044844 | Recruiting | Drug: Tofacitinib | Efficacy of Tofacitinib in the Systemic Sclerosis |
Bangabandhu Sheikh Mujib Medical University, Dhaka, Bangladesh |
November 2023 | Phase 2 |
| NCT04424303 | Recruiting | Drug: Tofacitinib | Ulcerative Colitis | Pfizer | December 4, 2020 | |
| NCT06278402 | Completed | Drug: Tofacitinib | Alopecia Areata Alopecia Totalis |
Jinnah Hospital | July 1, 2023 | Phase 3 |
") |
|
") |
 Frevecitinibum
Frevecitinibum
 Girocitinibum
Girocitinibum
 Envudeucitinibum
Envudeucitinibum
 CP-352664
CP-352664
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1





























 COA
COA
")
