
| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| 500mg |
|
||
| Other Sizes |
|

| 靶点 |
HIV-1/2 nucleotide reverse transcriptase
|
||
|---|---|---|---|
| 体外研究 (In Vitro) |
在MTT实验中,替诺福韦对HK-2细胞活力表现出细胞毒性作用,48小时和72小时的IC50值分别为2.77 μM。替诺福韦导致 HK-2 细胞的 ATP 水平下降。在 HK-2 细胞中,替诺福韦(3.0 至 28.8 μM)可增强蛋白质羰基化和氧化应激。此外,替诺福韦会导致HK-2细胞发生凋亡,这一过程是由线粒体损伤引起的[1]。当与 0.25% HEC 混合时,替诺福韦和 M48U1 会抑制激活的 PBMC 中 R5 向性 HIV-1BaL 和 X4 向性 HIV-1IIIb 的复制。此外,各种实验室毒株和患者来源的 HIV-1 分离株均受到抑制。 R5-tropic HIV-1BaL 的感染可通过 M48U1 和替诺福韦在 0.25% HEC 中的协同抗逆转录病毒作用来抑制,并且该制剂对 PBMC 无害[2]。
|
||
| 体内研究 (In Vivo) |
当给予 BLT 小鼠(20、50、140 或 300 mg/kg)时,富马酸替诺福韦二吡呋酯在 BLT 人源化小鼠中针对阴道 HIV 攻击表现出剂量依赖性功效。在 BLT 小鼠中,富马酸替诺福韦二吡呋酯(50、140 或 300 毫克/千克)可显着降低 HIV 传播[3]。在慢性 WHV 感染的土拨鼠中,富马酸替诺福韦二吡呋酯(0.5、1.5 或 5.0 mg/kg/天,口服)可导致血清病毒血症出现剂量依赖性下降。富马酸替诺福韦二吡呋酯治疗土拨鼠慢性 HBV 感染模型既安全又有效[4]。
|
||
| 细胞实验 |
细胞活力测定[2]
使用细胞计数试剂盒-8(CCK-8)测定细胞存活率。将小鼠NPC接种在96孔板上,密度为每孔1×104个细胞。孵育过夜后,用DMSO(0.55mg/ml,阴性对照)、胞嘧啶β-D-呋喃阿拉伯糖苷(Ara-C,7μg/ml,阳性对照)或不同浓度的抗逆转录病毒药物(0.1×、0.3×、0.5×和1×)处理细胞。每三天更换一半的培养基液体。在第2、4、6和8天,将10μl CCK-8溶液加入细胞培养的每个孔中,并将平板在37°C下再孵育2小时。然后使用微孔板读数器在450nm的吸光度下测量光密度。不含细胞的培养基作为空白对照。使用以下方程式计算细胞存活率:细胞存活率=(OD药物处理组-OD空白)/(OD DMSO处理组-OD-空白)。实验一式三份,独立重复至少三次。[2] 蛋白质印迹[2] 用M-PER蛋白提取缓冲液(Pierce)裂解小鼠NPC。使用二辛可宁酸(BCA)蛋白检测试剂盒(Pierce)测定总蛋白浓度。使用10%和15%凝胶进行分析性SDS-聚丙烯酰胺凝胶电泳(SDS-PAGE)。然后将蛋白质转移到免疫印迹聚偏二氟乙烯膜(Bio-Rad)上。在5%无脂牛奶中封闭1小时后,将膜与半胱氨酸天冬氨酸蛋白酶-3(1:1000;Cell Signaling Technologies)、聚ADP核糖聚合酶(PARP,1:1000;细胞信号技术)和肌动蛋白(1:5000;Sigma-Aldrich)的一抗在4°C下孵育过夜,然后在室温下与辣根过氧化物酶偶联的二抗孵育1小时。使用化学发光底物溶液检测蛋白质信号。每个条带的密度由Image Lab软件确定,并使用Image J程序进行分析。 |
||
| 动物实验 |
|
||
| 药代性质 (ADME/PK) |
Absorption, Distribution and Excretion
Following IV administration of tenofovir, approximately 70-80% of the dose is recovered in the urine as unchanged tenofovir within 72 hours of dosing. Following single dose, oral administration of tenofovir, the terminal elimination half-life of tenofovir is approximately 17 hours. After multiple oral doses of tenofovir 300 mg once daily (under fed conditions), 32 + or - 10% of the administered dose is recovered in urine over 24 hours. Tenofovir is eliminated by a combination of glomerular filtration and active tubular secretion. There may be competition for elimination with other compounds that are also renally eliminated. In vitro binding of tenofovir to human plasma or serum proteins is less than 0.7 and 7.2%, respectively, over the tenofovir concentration range 0.01 to 25 ug/mL. The volume of distribution at steady-state is 1.3 + or - 0.6 L/kg and 1.2 + or - 0.4 L/kg, following intravenous administration of tenofovir 1.0 mg/kg and 3.0 mg/kg. Viread is a water soluble diester prodrug of the active ingredient tenofovir. The oral bioavailability of tenofovir from Viread in fasted subjects is approximately 25%. Following oral administration of a single dose of Viread 300 mg to HIV-1 infected subjects in the fasted state, maximum serum concentrations (Cmax) are achieved in 1.0 + or - 0.4 hr. Cmax and AUC values are 0.30 + or - 0.09 ug/mL and 2.29 + or - 0.69 ug hr/mL, respectively. Administration of Viread 300 mg tablets following a high-fat meal (approximately 700 to 1000 kcal containing 40 to 50% fat) increases the oral bioavailability, with an increase in tenofovir AUC of approximately 40% and an increase in Cmax of approximately 14%. However, administration of Viread with a light meal did not have a significant effect on the pharmacokinetics of tenofovir when compared to fasted administration of the drug. Food delays the time to tenofovir Cmax by approximately 1 hour. Cmax and AUC of tenofovir are 0.33 + or - 0.12 ug/mL and 3.32 + or - 1.37 ug hr/mL following multiple doses of Viread 300 mg once daily in the fed state, when meal content was not controlled. For more Absorption, Distribution and Excretion (Complete) data for TENOFOVIR DISOPROXIL FUMARATE (6 total), please visit the HSDB record page. Metabolism / Metabolites Tenofovir disoproxil fumarate is a prodrug and is not active until it undergoes diester hydrolysis in vivo to tenofovir and subsequently is metabolized to the active metabolite (tenofovir diphosphate). Biological Half-Life Following single dose, oral administration of Viread, the terminal elimination half-life of tenofovir is approximately 17 hours. |
||
| 毒性/毒理 (Toxicokinetics/TK) |
Interactions
Potential pharmacokinetic interaction with drugs that reduce renal function or that may compete with tenofovir for active renal tubular secretion (i.e., acyclovir, cidofovir, ganciclovir, valacyclovir, valganciclovir); increased plasma concentrations of tenofovir or the concomitantly administered drug may occur. The manufacturer of tenofovir states that tenofovir should not be used with adefovir for the treatment of hepatitis B virus (HBV) infection. Pharmacokinetic interaction with atazanavir sulfate (decrease plasma concentrations and AUC of atazanavir (minimum concentration decreased 40%) and increased plasma concentrations and AUC of tenofovir when atazanavir 400 mg and tenofovir disoproxil fumarate 300 mg given once daily). Pharmacokinetic interaction with ritonavir-boosted atazanavir sulfate (decrease plasma concentrations and AUC of atazanavir (minimum concentration decreased 23%) and increased plasma concentrations and AUC of tenofovir when atazanavir 300 mg, ritonavir 100 mg, and tenofovir disoproxil fumarate 300 mg given once daily). If used concomitantly, a dosage regimen of atazanavir 300 mg, ritonavir 100 mg, and tenofovir disoproxil fumarate 300 mg given once daily with food is recommended; atazanavir should not be used with tenofovir unless low-dose ritonavir is a component of the regimen. Monitor for tenofovir toxicity and discontinue the drug if tenofovir-associated adverse effects occur. If atazanavir is used concomitantly with tenofovir and a histamine H2-receptor antagonist, the recommended dosage for treatment-experienced patients is atazanavir 400 mg, ritonavir 100 mg, and tenofovir disoproxil fumarate 300 mg given once daily with food. Pharmacokinetic interaction with the buffered didanosine preparation (pediatric oral solution admixed with antacid; Videx) or delayed-release capsules containing enteric-coated pellets of didanosine (Videx EC) resulting in increased plasma concentrations and AUC of didanosine; no change in tenofovir pharmacokinetics. Potential for early virologic failure, rapid selection of resistant mutations, immunologic nonresponse (e.g., decline in CD4+ T-cell count), and increased risk of didanosine-associated adverse effects (e.g., pancreatitis, neuropathy). Caution is advised if didanosine and tenofovir are used concomitantly and patients should be monitored closely for didanosine-associated adverse effects; didanosine should be discontinued if such effects occur. If didanosine delayed-release capsules are used with tenofovir disoproxil fumarate, the recommended dosage of didanosine is 250 mg once daily for those weighing 60 kg or more with creatinine clearances of 60 mL/minute or greater and 200 mg once daily for those weighing less than 60 kg with creatinine clearances of 60 mL/minute or greater. Didanosine delayed-release capsules and tenofovir may be taken at the same time with a light meal (no more than 400 kcal, no more than 20% fat) or in the fasted state. For more Interactions (Complete) data for TENOFOVIR DISOPROXIL FUMARATE (10 total), please visit the HSDB record page. |
||
| 参考文献 |
|
||
| 其他信息 |
Therapeutic Uses
Anti-HIV Agents, Reverse Transcriptase Inhibitors Tenofovir disoproxil fumarate is used in conjunction with other antiretroviral agents for the treatment of human immunodeficiency virus type 1 (HIV-1) infections in adults. /Included in US product labeling/ Tenofovir is used for the management of chronic hepatitis B virus (HBV) infection in adults. This indication is based on histologic, virologic, biochemical, and serologic responses in adults with hepatitis B e antigen (HBeAg)-positive or -negative chronic HBV with compensated liver function. Tenofovir disoproxil fumarate (TDF), emtricitabine (FTC), and efavirenz (EFV) are the three components of the once-daily, single tablet regimen (Atripla) for treatment of HIV-1 infection. Previous cell culture studies have demonstrated that the double combination of tenofovir (TFV), the parent drug of TDF, and FTC were additive to synergistic in their anti-HIV activity, which correlated with increased levels of intracellular phosphorylation of both compounds. In this study, /researchers/ demonstrated the combinations of TFV+FTC, TFV+EFV, FTC+EFV, and TFV+FTC+EFV synergistically inhibit HIV replication in cell culture and synergistically inhibit HIV-1 reverse transcriptase (RT) catalyzed DNA synthesis in biochemical assays. Several different methods were applied to define synergy including median-effect analysis, MacSynergyII and quantitative isobologram analysis. We demonstrated that the enhanced formation of dead-end complexes (DEC) by HIV-1 RT and TFV-terminated DNA in the presence of FTC-triphosphate (TP) could contribute to the synergy observed for the combination of TFV+FTC, possibly through reduced terminal NRTI excision. Furthermore, /researchers/ showed that EFV facilitated efficient formation of stable, DEC-like complexes by TFV- or FTC-monophosphate (MP)-terminated DNA and this can contribute to the synergistic inhibition of HIV-1 RT by TFV-diphosphate (DP)+EFV and FTC-TP+EFV combinations. This study demonstrated a clear correlation between the synergistic antiviral activities of TFV+FTC, TFV+EFV, FTC+EFV, and TFV+FTC+EFV combinations and synergistic HIV-1 RT inhibition at the enzymatic level. /Researchers/ propose the molecular mechanisms for the TFV+FTC+EFV synergy to be a combination of increased levels of the active metabolites TFV-DP and FTC-TP and enhanced DEC formation by a chain-terminated DNA and HIV-1 RT in the presence of the second and the third drug in the combination. This study furthers the understanding of the longstanding observations of synergistic anti-HIV-1 effects of many NRTI+NNRTI and certain NRTI+NRTI combinations in cell culture, and provides biochemical evidence that combinations of anti-HIV agents can increase the intracellular drug efficacy, without increasing the extracellular drug concentrations. Drug Warnings /BOXED WARNING/ WARNING: LACTIC ACIDOSIS/SEVERE HEPATOMEGALY WITH STEATOSIS and POST TREATMENT EXACERBATION OF HEPATITIS. Lactic acidosis and severe hepatomegaly with steatosis, including fatal cases, have been reported with the use of nucleoside analogs, including Viread, in combination with other antiretrovirals. Severe acute exacerbations of hepatitis have been reported in HBV-infected patients who have discontinued anti-hepatitis B therapy, including Viread. Hepatic function should be monitored closely with both clinical and laboratory follow-up for at least several months in patients who discontinue anti-hepatitis B therapy, including Viread. If appropriate, resumption of anti-hepatitis B therapy may be warranted. Lactic acidosis and severe hepatomegaly with steatosis (sometimes fatal) have been reported rarely in patients receiving nucleoside reverse transcriptase inhibitors alone or in conjunction with other antiretroviral agents. Most reported cases have involved women; obesity and long-term therapy with a nucleoside reverse transcriptase inhibitor also may be risk factors. Caution should be observed when nucleoside analogs are used in patients with known risk factors for liver disease; however, lactic acidosis and severe hepatomegaly with steatosis have been reported in patients with no known risk factors. Tenofovir therapy should be interrupted in any patient with clinical or laboratory findings suggestive of lactic acidosis or pronounced hepatotoxicity (signs of hepatotoxicity include hepatomegaly and steatosis even in the absence of marked increases in serum aminotransferase concentrations). Redistribution or accumulation of body fat, including central obesity, dorsocervical fat enlargement (buffalo hump), peripheral wasting, facial wasting, breast enlargement, and general cushingoid appearance, has been reported with antiretroviral therapy. The most common adverse effects in HIV-infected patients receiving tenofovir disoproxil fumarate are rash, diarrhea, headache, pain, depression, asthenia, and nausea. The most common adverse effect in HIV-infected patients receiving tenofovir disoproxil fumarate is nausea. For more Drug Warnings (Complete) data for TENOFOVIR DISOPROXIL FUMARATE (14 total), please visit the HSDB record page. |
| 分子式 |
C19H30N5O10P.C4H4O4
|
|
|---|---|---|
| 分子量 |
635.51
|
|
| 精确质量 |
635.183
|
|
| 元素分析 |
C, 43.47; H, 5.39; N, 11.02; O, 35.24; P, 4.87
|
|
| CAS号 |
202138-50-9
|
|
| 相关CAS号 |
Tenofovir Disoproxil;201341-05-1;Tenofovir;147127-20-6;Tenofovir maleate;1236287-04-9; 201341-05-1 (free); 202138-50-9 (fumarate); 206184-49-8 (hydrate); 379270-37-8 (alafenamide); 1571075-19-8 (aspartate)
|
|
| PubChem CID |
6398764
|
|
| 外观&性状 |
White, fine, powder-like crystals
|
|
| 密度 |
1.45 g/cm3
|
|
| 沸点 |
642.7ºC at 760 mmHg
|
|
| 熔点 |
219ºC
|
|
| 闪点 |
342.5ºC
|
|
| 蒸汽压 |
2.06E-16mmHg at 25°C
|
|
| LogP |
3.328
|
|
| tPSA |
269.85
|
|
| 氢键供体(HBD)数目 |
3
|
|
| 氢键受体(HBA)数目 |
18
|
|
| 可旋转键数目(RBC) |
19
|
|
| 重原子数目 |
43
|
|
| 分子复杂度/Complexity |
817
|
|
| 定义原子立体中心数目 |
1
|
|
| SMILES |
P(C([H])([H])O[C@]([H])(C([H])([H])[H])C([H])([H])N1C([H])=NC2=C(N([H])[H])N=C([H])N=C12)(=O)(OC([H])([H])OC(=O)OC([H])(C([H])([H])[H])C([H])([H])[H])OC([H])([H])OC(=O)OC([H])(C([H])([H])[H])C([H])([H])[H].O([H])C(C([H])=C([H])C(=O)O[H])=O
|
|
| InChi Key |
VCMJCVGFSROFHV-WZGZYPNHSA-N
|
|
| InChi Code |
InChI=1S/C19H30N5O10P.C4H4O4/c1-12(2)33-18(25)28-9-31-35(27,32-10-29-19(26)34-13(3)4)11-30-14(5)6-24-8-23-15-16(20)21-7-22-17(15)24;5-3(6)1-2-4(7)8/h7-8,12-14H,6,9-11H2,1-5H3,(H2,20,21,22);1-2H,(H,5,6)(H,7,8)/b;2-1+/t14-;/m1./s1
|
|
| 化学名 |
9-((R)-2-((Bis(((isopropoxycarbonyl)oxy)methoxy)phosphinyl)methoxy)propyl)adenine, fumarate
|
|
| 别名 |
|
|
| HS Tariff Code |
2934.99.9001
|
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month 注意: 请将本产品存放在密封且受保护的环境中,避免吸湿/受潮。 |
|
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
|
|||
|---|---|---|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 2.5 mg/mL (3.93 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 25.0 mg/mL澄清DMSO储备液加入到400 μL PEG300中,混匀;然后向上述溶液中加入50 μL Tween-80,混匀;加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: ≥ 2.5 mg/mL (3.93 mM) (饱和度未知) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 25.0 mg/mL澄清DMSO储备液加入900 μL 20% SBE-β-CD生理盐水溶液中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 View More
配方 3 中的溶解度: ≥ 2.5 mg/mL (3.93 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 配方 4 中的溶解度: 20 mg/mL (31.47 mM) in PBS (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液; 超声助溶. 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.5735 mL | 7.8677 mL | 15.7354 mL | |
| 5 mM | 0.3147 mL | 1.5735 mL | 3.1471 mL | |
| 10 mM | 0.1574 mL | 0.7868 mL | 1.5735 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
|
|---|
|
 NNRT-IN-5
NNRT-IN-5
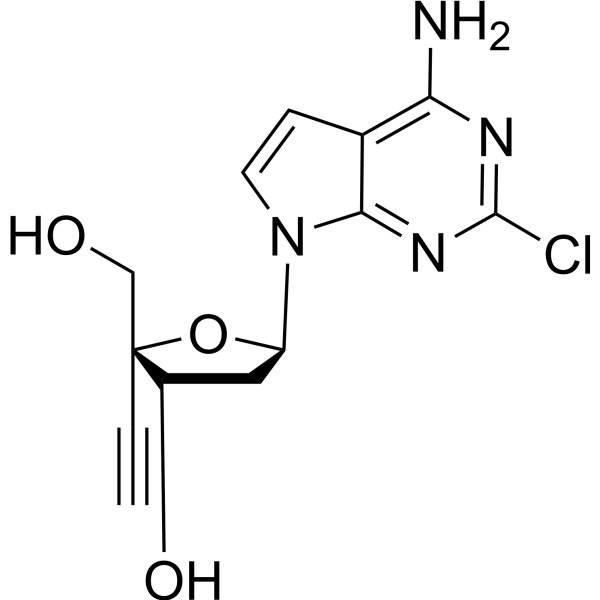 MK-8527
MK-8527
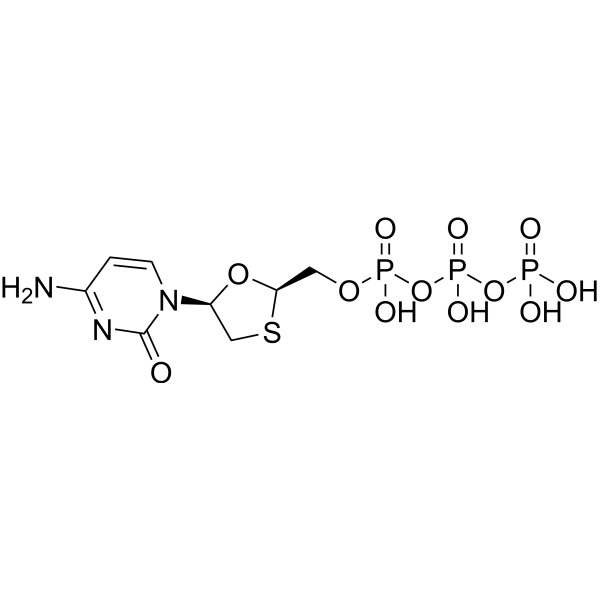 Lamivudine triphosphate
Lamivudine triphosphate
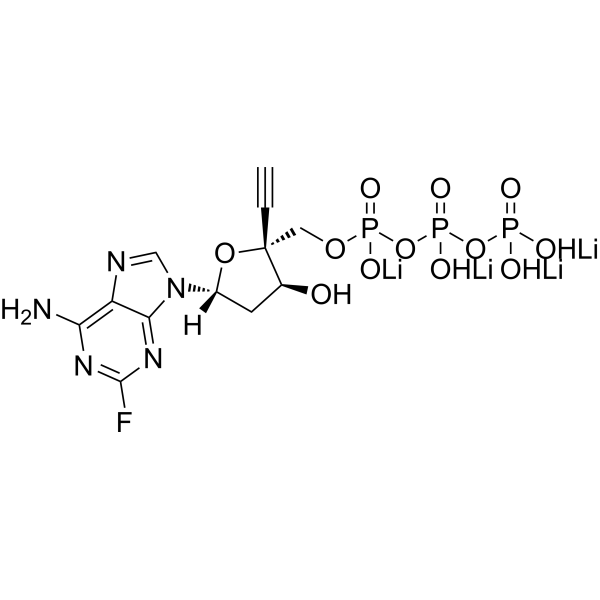 EFdA-TP tetralithium
EFdA-TP tetralithium
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1





























 COA
COA


