
| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 1mg |
|
||
| 5mg |
|
||
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| 500mg |
|
||
| 1g |
|
||
| 5g |
|
||
| Other Sizes |
|
| 靶点 |
HIV-1/2 nucleotide reverse transcriptase
|
||
|---|---|---|---|
| 体外研究 (In Vitro) |
替诺福韦艾拉酚胺 (GS-7340) 在所有细胞类型中的抗病毒活性范围为 5 至 7 nM,但 MT-4 和 MT-2 细胞的 CC50 范围为 4.7 至 42 μM。一组 HIV-1 和 HIV-2 分离株(包括 HIV-1 M 组亚型 A 至 G 以及 N 组和 O 组分离株)用于评估 TAF 的抗病毒活性。 TAF EC50 平均值为 3.5 nM,范围为 0.1 至 12 nM。相比之下,对于在 PBMC 中评估的 29 种主要 HIV-1 毒株,AZT(内部对照)的平均 EC50 为 11.8 nM。 TAF 对 HIV-2 分离株的平均 EC50 为 1.8 nM,而 AZT 的平均 EC50 为 6.4 nM [2]。
|
||
| 体内研究 (In Vivo) |
与富马酸替诺福韦二吡呋酯 (TDF) 相比,替诺福韦的酰胺化前药替诺福韦艾拉酚胺 (GS-7340) 半富马酸盐具有更好的口服生物利用度和血浆稳定性 [1]。
|
||
| 酶活实验 |
肠和肝S9稳定性[1]
将GS-7340与狗或人的肠道和肝脏S9级分在10μM下在37°C下以96孔板型孵育120分钟。在添加化合物后的指定时间点,用9体积的含有25%乙腈和50%甲醇的水溶液淬灭样品。将平板在3000g下离心30分钟,并通过LC–MS/MS分析10μL所得溶液。数据(分析物与内标峰面积之比)以半对数标度绘制,并使用指数拟合进行拟合。假设一阶动力学,确定半衰期和代谢速率。使用充分搅拌的肝脏清除模型,通过已报道的方法从半衰期计算预测的肝脏提取。 |
||
| 细胞实验 |
Caco-2渗透率[1]
如先前报道的,使用接种在12孔板中的人结肠癌细胞系caco-2的融合单层进行双向渗透性研究。研究了浓度(10、100或1000μM)或外排转运体抑制对GS-7340渗透的影响。在细胞单层与10μM环孢菌素a(CsA)在转运缓冲液中预孵育30分钟以使转运蛋白结合位点饱和后,评估包括P-糖蛋白(Pgp)在内的外排转运蛋白的抑制作用。预培养后,加入含有CsA和GS-7340的新鲜测定缓冲液,并开始测定。每种测定重复进行,并测定对照化合物(阿替洛尔、普萘洛尔和地高辛)的渗透性,以满足每批测定板的验收标准。 |
||
| 动物实验 |
|
||
| 药代性质 (ADME/PK) |
Absorption, Distribution and Excretion
As compared to the parent molecule, [tenofovir], tenofovir alafenamide presents a lipophilic group that masks the negative charge of the parent moiety which improves its oral bioavailability. Tenofovir alafenamide is highly stable in plasma and, after administration of this prodrug, there is a low concentration of tenofovir in plasma. After oral administration, tenofovir alafenamide is rapidly absorbed by the gut. When a single dose is administered, a peak concentration of 16 ng/ml of the parent compound, corresponding to about 73% of the dose, is observed after 2 hours with an AUC of 270 ng\*h/mL. Once inside the body, tenofovir alafenamide enters hepatocytes by passive diffusion regulated by the organic anion transporters 1B1 and 1B3 for its activation. Administration of tenofovir alafenamide concomitantly with a high-fat meal results in an increase of about 65% in its internal exposure. Tenofovir alafenamide has been registered to present a bile elimination that corresponds to 47% of the administered dose and a renal elimination the represents about 36%. From the recovered dose in urine, about 75% is represented as unchanged [tenofovir] followed by uric acid and a small dose of tenofovir alafenamide. On the other hand, in feces, 99% of the recovered dose corresponds to tenofovir. In clinical trials, the reported volume of distribution of tenofovir alafenamide was higher than 100 L. The reported clearance rate of tenofovir alafenamide is 117 L/h. In patients with severe renal impairment, this value can be decreased by 50%, reporting a rate of 61.7 L/h. Metabolism / Metabolites To be activated, tenofovir alafenamide is required to be hydrolyzed to the parent compound [tenofovir] by the activity of cathepsin A or carboxylesterase 1. Tenofovir alafenamide presents significant plasma stability and hence, its activation is performed inside the target cells. After activation, tenofovir is further processed and after 1-2 days, it is detected in plasma almost completely transformed to uric acid. Biological Half-Life The reported half-life for tenofovir alafenamide is of 0.51 hours. |
||
| 毒性/毒理 (Toxicokinetics/TK) |
Protein Binding
Tenofovir alafenamide is reported to bind to plasma proteins and _ex vivo_ studies have registered that approximately 80% of the administered dose of this drug is presented in a bound state. |
||
| 参考文献 |
|
||
| 其他信息 |
Pharmacodynamics
Tenofovir alafenamide has been shown to be a potent inhibitor of hepatitis B viral replication. Tenofovir alafenamide presents a better renal tolerance when compared with the counterpart [tenofovir disoproxil]. This improved safety profile seems to be related to a lower plasma concentration of tenofovir. In clinical trials, tenofovir alafenamide was shown to present 5-fold more potent antiviral activity against HIV-1 when compared to tenofovir disoproxil. |
| 分子式 |
C21H29N6O5P
|
|
|---|---|---|
| 分子量 |
476.47
|
|
| 精确质量 |
476.193
|
|
| 元素分析 |
C, 52.94; H, 6.13; N, 17.64; O, 16.79; P, 6.50
|
|
| CAS号 |
379270-37-8
|
|
| 相关CAS号 |
Tenofovir alafenamide fumarate;379270-38-9;Tenofovir alafenamide hemifumarate;1392275-56-7
|
|
| PubChem CID |
9574768
|
|
| 外观&性状 |
White to off-white solid
|
|
| 密度 |
1.39±0.1 g/cm3
|
|
| 沸点 |
640.4±65.0 °C at 760 mmHg
|
|
| 熔点 |
104-107 ºC
|
|
| 闪点 |
341.1±34.3 °C
|
|
| 蒸汽压 |
0.0±1.9 mmHg at 25°C
|
|
| 折射率 |
1.630
|
|
| LogP |
2.2
|
|
| tPSA |
153.29
|
|
| 氢键供体(HBD)数目 |
2
|
|
| 氢键受体(HBA)数目 |
10
|
|
| 可旋转键数目(RBC) |
12
|
|
| 重原子数目 |
33
|
|
| 分子复杂度/Complexity |
680
|
|
| 定义原子立体中心数目 |
3
|
|
| SMILES |
[P@](C([H])([H])OC([H])(C([H])([H])[H])C([H])([H])N1C([H])=NC2=C(N([H])[H])N=C([H])N=C12)(N([H])C([H])(C(=O)OC([H])(C([H])([H])[H])C([H])([H])[H])C([H])([H])[H])(=O)OC1C([H])=C([H])C([H])=C([H])C=1[H]
|
|
| InChi Key |
LDEKQSIMHVQZJK-AZFZMOAFSA-N
|
|
| InChi Code |
InChI=1S/C21H29N6O5P/c1-14(2)31-21(28)16(4)26-33(29,32-17-8-6-5-7-9-17)13-30-15(3)10-27-12-25-18-19(22)23-11-24-20(18)27/h5-9,11-12,14-16H,10,13H2,1-4H3,(H,26,29)(H2,22,23,24)/t15-,16+,33?/m1/s1
|
|
| 化学名 |
(S)-isopropyl 2-(((S)-((((R)-1-(6-amino-9H-purin-9-yl)propan-2-yl)oxy)methyl)(phenoxy)phosphoryl)amino)propanoate
|
|
| 别名 |
|
|
| HS Tariff Code |
2934.99.9001
|
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
|
|||
|---|---|---|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 2.08 mg/mL (4.37 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 20.8 mg/mL澄清DMSO储备液加入400 μL PEG300中,混匀;然后向上述溶液中加入50 μL Tween-80,混匀;加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: ≥ 2.08 mg/mL (4.37 mM) (饱和度未知) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 20.8 mg/mL澄清DMSO储备液加入900 μL 20% SBE-β-CD生理盐水溶液中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 View More
配方 3 中的溶解度: ≥ 2.08 mg/mL (4.37 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 配方 4 中的溶解度: 40 mg/mL (83.95 mM) in PBS (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液; 超声助溶. 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.0988 mL | 10.4938 mL | 20.9877 mL | |
| 5 mM | 0.4198 mL | 2.0988 mL | 4.1975 mL | |
| 10 mM | 0.2099 mL | 1.0494 mL | 2.0988 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
|
|---|
|
 NNRT-IN-5
NNRT-IN-5
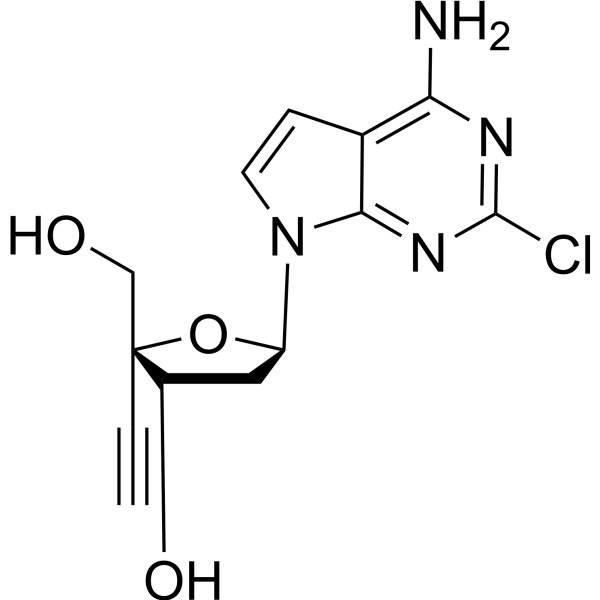 MK-8527
MK-8527
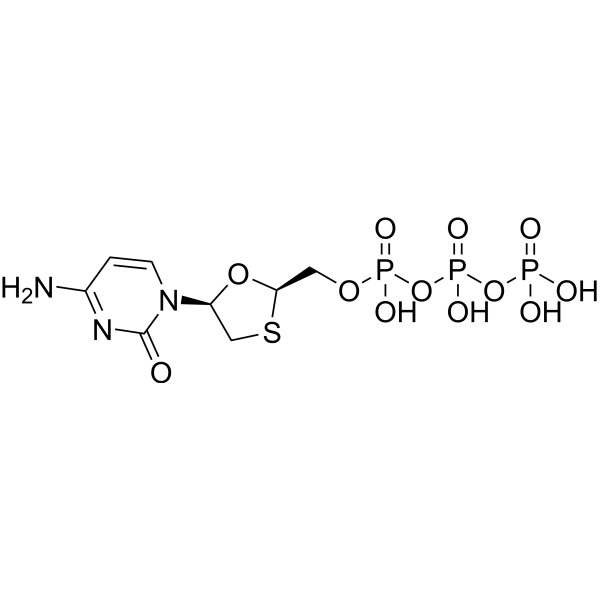 Lamivudine triphosphate
Lamivudine triphosphate
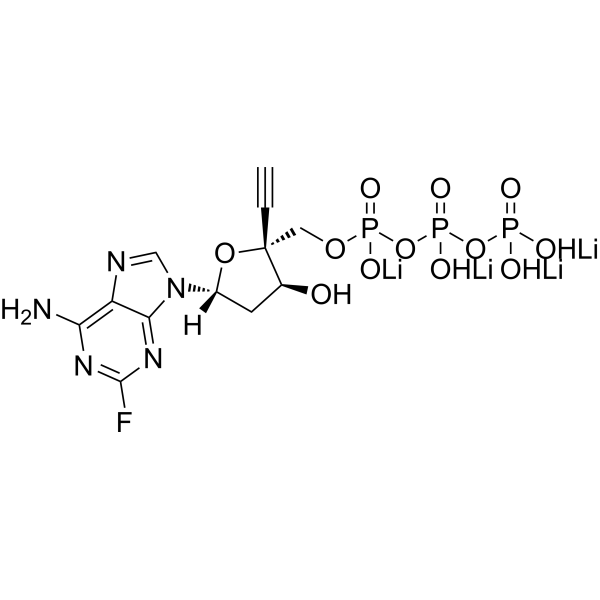 EFdA-TP tetralithium
EFdA-TP tetralithium
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1






























 COA
COA
")
")
