| 规格 | 价格 | |
|---|---|---|
| 500mg | ||
| 1g | ||
| Other Sizes |
| 靶点 |
Prodrug of ganetespib
|
|---|---|
| 体外研究 (In Vitro) |
持续的HSP90抑制导致体外客户端蛋白的持久不稳定;然而,短暂暴露需要>10倍的药物才能产生类似的效果[1]。
|
| 体内研究 (In Vivo) |
在体内,KIT在单剂量HSP90i后迅速降解,但在一天内恢复到基线水平。HSP90水平在HSP90i暴露16小时后增加并稳定,在随后的近期暴露后没有升高,提供了稳定蛋白质的伴侣蛋白的功能库,并为HSP90i再暴露提供了更大的治疗活性的手段。在KIT或EGFR驱动的小鼠肿瘤模型中,与D1治疗相比,在第1天和第2天给予的HSP90i(D1/D2)显示出增加的生物活性。在一项MCT犬试验中,与D1和D1/D4方案相比,D1/D2给药HSP90i与KIT持续下调、50%的客观有效率和100%的临床获益率相关。
|
| 酶活实验 |
HSP90结合测定。[1]
H1975细胞在RPMI-1640和10%FBS中培养,以每孔3×105个细胞的密度接种在6孔板中。24小时后,按指示用ganetespib处理细胞,并在37°C下孵育。细胞在冷PBS中洗涤两次,然后在冷HSP90结合缓冲液(20mM HEPES pH 7.3、1mM EDTA、100mM KCl、5mM MgCl、0.01%v/v NP-40、0.5mg/mL牛丙种球蛋白、1mM TCEP)中裂解,在冰上孵育10分钟,然后进行三次冷冻/解冻循环。通过在14000 x g下离心来澄清裂解物。为了去除未结合的ganetespib,将裂解物通过40K MWCO尺寸的排除柱。为了滴定未被占据的HSP90结合位点,将10μM的氘化形式的甘尼特spib(D3甘尼特Spib)添加到洗脱液中,并在4°C下孵育2小时,然后通过尺寸排除柱以去除未结合的D3甘尼特斯pib。通过BCA蛋白质测定法对流经的总蛋白质进行定量,并将所有样品稀释至1 mg/mL。通过LC/MS-MS测量ganetespib和D3的浓度。使用Phenomenex Kinetex柱(C18,30×2.1 mm,2.6µm),每个样品的运行时间为3.5分钟。使用以下方程来计算与HSP90结合的ganetespib的百分比(HSP90占有率):[ganetespib]/([ganetespib]+[D3-ganetespip])x100[1]。 |
| 细胞实验 |
蛋白质印迹[1]
在体外测定之后,将肿瘤细胞在冰上的裂解缓冲液中破坏10分钟。为了进行药效学分析,切除异种移植物肿瘤(平均体积为100-200 mm3),切成两半,并在液氮中快速冷冻。使用FastPrep-24匀浆器和裂解基质a将每个肿瘤片段裂解在0.5mL裂解缓冲液中。通过离心澄清裂解物,并在转移到硝化纤维素膜之前通过SDS-PAGE解析等量的蛋白质。用起始阻断T20阻断缓冲液阻断膜,并用指示的抗体进行免疫印迹。抗体-抗原复合物使用奥德赛系统(LI-COR)进行可视化。 |
| 动物实验 |
Female CB.17 (SCID) mice at 7–12 weeks of age were maintained in a pathogen-free environment and all in vivo procedures were approved by the Synta Pharmaceuticals Corp. Institutional Animal Care and Use Committee. Human GIST882 cells were provided by Dr. Jonathan Fletcher (Dana Farber Cancer Institute) and implanted subcutaneously at 10 × 106 into mice. Mice bearing established tumors (~110 mm3) were randomized into treatment groups of 8 and dosed intravenously with vehicle or ganetespib, formulated in DRD (10% DMSO, 18% Cremophor RH 40, 3.6% dextrose), using the schedules indicated. Human H1975 NSCLC cells were purchased from the ATCC, selected to stably express a HIF-1α-LUC reporter and implanted at 10 × 106 into mice. Mice bearing established tumors (~143 mm3) were randomized into treatment groups of 4 and dosed intravenously with vehicle or ganetespib, formulated in DRD, using the schedules indicated. Tumor volumes (V) were calculated by caliper measurements of the width (W), length (L) and thickness (T) of each tumor using the formula: V=0.5236(LWT). Tumor growth inhibition was determined as described previously.[1]
|
| 药代性质 (ADME/PK) |
Absorption, Distribution and Excretion
Bioavailability is 58-70% following oral administration, compared to parenteral forms (i.v. and i.m. = 100%). Biological Half-Life t1/2-alpha = minutes (10-20 minutes in rat); t1/2-beta = a few hours |
| 毒性/毒理 (Toxicokinetics/TK) |
Hepatotoxicity
Defibrotide therapy has not been linked to serum aminotransferase elevations or with instances of clinically apparent liver injury separate from the features of SOS for which it is given. In a trial of defibrotide as prophylaxis against SOS conducted in 356 children undergoing HCT, rates of severe adverse events such as hemorrhage, gastrointestinal complaints and liver injury were similar in those receiving defibrotide as in untreated children. Likelihood score: E (unlikely cause of clinically apparent acute liver injury). |
| 参考文献 |
[1]. Consecutive Day HSP90 Inhibitor Administration Improves Efficacy in Murine Models of KIT-Driven Malignancies and Canine Mast Cell Tumors. Clin Cancer Res. 2018 Dec 15;24(24):6396-6407.
|
| 其他信息 |
Defibrotide is the sodium salt of a mixture of single-stranded oligodeoxyribonucleotides derived from porcine mucosal DNA. It has been shown to have antithrombotic, anti-inflammatory and anti-ischemic properties (but without associated significant systemic anticoagulant effects). It is marketed under the brand names Dasovas (FM), Noravid, and Prociclide in a variety of countries. In the USA it is was approved in March, 2016 as Defitelio.
Defibrotide is a complex mixture of single stranded polydeoxyribonucleotides derived from porcine intestinal mucosa that has antithrombotic and profibrinolytic activity and is used in the treatment of severe sinusoidal obstruction syndrome (SOS) after hematopoietic cell transplantation (HCT). Defibrotide is used in patients with severe liver injury and has not been associated with worsening of serum aminotransferase elevations during therapy and has not been linked to cases of clinically apparent, idiosyncratic liver injury. Defibrotide is a mixture of single-stranded oligodeoxyribonucleotides derived from the intestinal mucosa of pigs, with anti-thrombotic, thrombolytic, and fibrinolytic activities. Upon administration, and although the exact mechanism of action has yet to be fully elucidated, defibrotide induces the release of prostaglandin I2 (PGI2), E2 (PGE2), and prostacyclin and reduces the expression of adhesion molecules on endothelial cells. This relaxes the smooth muscle of blood vessels and prevents platelets from adhering to each other and to the endothelium. This protects the endothelium lining bloods vessels. Defibrotide increases tissue plasminogen activator (t-PA) and decreases plasminogen activator inhibitor-1 activity. This increases the activity of plasmin, prevents blood clot formation and dissolves blood clots. See also: Defibrotide Sodium (annotation moved to). Drug Indication Indicated for the treatment of severe hepatic veno-occlusive disease (VOD), also known as sinusoidal obstruction syndrome (SOS), with renal or pulmonary dysfunction following hematopoietic stem-cell transplantation (HSCT). FDA Label Mechanism of Action The drug appears to prevent the formation of blood clots and to help dissolve blood clots by increasing levels of prostaglandin I2, E2, and prostacyclin, altering platelet activity, increasing tissue plasminogen activator function, and decreasing activity of tissue plasminogen activator inhibitor. Prostaglandin I2 relaxes the smooth muscle of blood vessels and prevents platelets from adhering to each other. Prostaglandin E2 at certain concentrations also inhibits platelet aggregation. Moreover, the drug provides additional beneficial anti-inflammatory and antiischemic activities as recent sudies have shown. It is yet unclear, if the latter effects can be utilized clinically (e.g., treatment of ischemic stroke). Pharmacodynamics Defibrotide is a deoxyribonucleic acid derivative extracted from mammalian organs, which has been developed for the treatment of a number of vascular disorders. It appears to increase fibrinolysis and may possess antithrombotic, antiatherosclerotic and anti-ischaemic actions, probably due to its ability to selectively increase prostaglandin I2 and E2 levels and to increase tissue plasminogen activator and decrease plasminogen activator inhibitor function. Defibrotide is available as an intravenous and intramuscular preparation, and also as an oral formulation for long term use. |
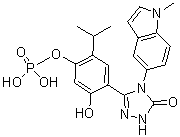
| 分子式 |
C20H21N4O6P
|
|---|---|
| 分子量 |
444.378
|
| 精确质量 |
444.12
|
| 元素分析 |
C, 54.06; H, 4.76; N, 12.61; O, 21.60; P, 6.97
|
| CAS号 |
1118915-78-8
|
| 相关CAS号 |
1402907-09-8 (disodium);1118915-79-9 (monosodium);1118915-78-8 (free acid);83712-60-1(sodium);
|
| PubChem CID |
135565962
|
| 外观&性状 |
Typically exists as solid at room temperature
|
| LogP |
1.987
|
| tPSA |
152.41
|
| 氢键供体(HBD)数目 |
4
|
| 氢键受体(HBA)数目 |
7
|
| 可旋转键数目(RBC) |
5
|
| 重原子数目 |
31
|
| 分子复杂度/Complexity |
773
|
| 定义原子立体中心数目 |
0
|
| SMILES |
CC(C)C1=CC(=C(C=C1OP(=O)(O)O)O)C2=NN=C(N2C3=CC=C4C(=C3)C=CN4C)O
|
| InChi Key |
JNWFIPVDEINBAI-UHFFFAOYSA-N
|
| InChi Code |
InChI=1S/C20H21N4O6P/c1-11(2)14-9-15(17(25)10-18(14)30-31(27,28)29)19-21-22-20(26)24(19)13-4-5-16-12(8-13)6-7-23(16)3/h4-11,25H,1-3H3,(H,22,26)(H2,27,28,29)
|
| 化学名 |
5-hydroxy-2-isopropyl-4-(4-(1-methyl-1H-indol-5-yl)-5-oxo-4,5-dihydro-1H-1,2,4-triazol-3-yl)phenyl dihydrogen phosphate
|
| 别名 |
STA-1474; ganetespib-prodrug; STA 1474; STA-9090 prodrug; STA1474; STA9090 prodrug
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples
|
|---|---|
| 溶解度 (体内实验) |
注意: 如下所列的是一些常用的体内动物实验溶解配方,主要用于溶解难溶或不溶于水的产品(水溶度<1 mg/mL)。 建议您先取少量样品进行尝试,如该配方可行,再根据实验需求增加样品量。
注射用配方
注射用配方1: DMSO : Tween 80: Saline = 10 : 5 : 85 (如: 100 μL DMSO → 50 μL Tween 80 → 850 μL Saline)(IP/IV/IM/SC等) *生理盐水/Saline的制备:将0.9g氯化钠/NaCl溶解在100 mL ddH ₂ O中,得到澄清溶液。 注射用配方 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (如: 100 μL DMSO → 400 μL PEG300 → 50 μL Tween 80 → 450 μL Saline) 注射用配方 3: DMSO : Corn oil = 10 : 90 (如: 100 μL DMSO → 900 μL Corn oil) 示例: 以注射用配方 3 (DMSO : Corn oil = 10 : 90) 为例说明, 如果要配制 1 mL 2.5 mg/mL的工作液, 您可以取 100 μL 25 mg/mL 澄清的 DMSO 储备液,加到 900 μL Corn oil/玉米油中, 混合均匀。 View More
注射用配方 4: DMSO : 20% SBE-β-CD in Saline = 10 : 90 [如:100 μL DMSO → 900 μL (20% SBE-β-CD in Saline)] 口服配方
口服配方 1: 悬浮于0.5% CMC Na (羧甲基纤维素钠) 口服配方 2: 悬浮于0.5% Carboxymethyl cellulose (羧甲基纤维素) 示例: 以口服配方 1 (悬浮于 0.5% CMC Na)为例说明, 如果要配制 100 mL 2.5 mg/mL 的工作液, 您可以先取0.5g CMC Na并将其溶解于100mL ddH2O中,得到0.5%CMC-Na澄清溶液;然后将250 mg待测化合物加到100 mL前述 0.5%CMC Na溶液中,得到悬浮液。 View More
口服配方 3: 溶解于 PEG400 (聚乙二醇400) 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.2503 mL | 11.2516 mL | 22.5033 mL | |
| 5 mM | 0.4501 mL | 2.2503 mL | 4.5007 mL | |
| 10 mM | 0.2250 mL | 1.1252 mL | 2.2503 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1


































