| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 100mg |
|
||
| 250mg |
|
||
| 500mg |
|
||
| 1g |
|
||
| 2g |
|
||
| 5g |
|
||
| Other Sizes |
|

| 靶点 |
HCV ( EC50 = 92±5 nM )
|
||
|---|---|---|---|
| 体外研究 (In Vitro) |
体外活性:作为 HCV NS5B 聚合酶抑制剂,PSI-7977 对 HCV RNA 复制表现出比 PSI-7976 更有效的抑制活性,EC50 为 92 nM vs 1.07 μM,EC90 为 0.29 μM vs 2.99 μM,与孵育克隆 A 细胞一致与 PSI-7977 孵育相比,与 PSI-7976 孵育的克隆 A 细胞相比,PSI-7409 浓度更高。 PSI-7977 是 CatA 形成 PSI-352707 的有效底物,其效力是 PSI-7976 的 18-30 倍。然而,与 GS-7976 不同的是,CES1 介导的 PSI-7977 水解并不以时间依赖性方式进行。 S282T NS5B 聚合酶突变而非 S96T 突变赋予 PSI-7977 抗性,EC90 从 0.42 μM 增加至 7.8 μM。在 8 天细胞毒性测定中进行评估时,即使浓度高达 100 μM,PSI-7977 对 Huh7、HepG2、BxPC3 和 CEM 细胞也没有显示出细胞毒性。 PSI-7977 处理 14 天显示,在 HepG2 细胞中抑制 mtDNA 和 rDNA 的 IC90 分别为 72.1 μM 和 68.6 μM。 PSI-7977 对基因型 (GT) 1a、1b 和 2a(菌株 JFH-1)复制子以及含有 GT 2a(菌株 J6)、2b 和 3a NS5B 聚合酶的嵌合复制子表现出有效的活性。 JFH-1 NS5B 区域的序列分析表明,在 S282T 出现之前和之后选择了额外的氨基酸变化,包括 T179A、M289L、I293L、M434T 和 H479P,这是赋予 PSI-7977 抗性所必需的。细胞测定:将细胞(Huh7、HepG2、BxPC3 和 CEM)暴露于不同浓度的 PSI-7977 中 8 天。在生长期结束时,将来自 CellTiter 96 AQueous One Solution 细胞增殖测定试剂盒的 MTS 染料添加到每个孔中,并将板再孵育 2 小时。使用仅培养基对照孔作为空白,用 Victor3 读板器读取 490 nm 处的吸光度。通过比较含有细胞和 PSI-7977 的孔与未处理的细胞对照孔中的吸光度来确定 50% 抑制值 (IC50)。
|
||
| 体内研究 (In Vivo) |
440 mg/kg/d 治疗组和 44 mg/kg/d 治疗组的人源化肝脏小鼠的平均血浆 ALT 水平低于正常上限,并且与媒介物治疗的人源化肝脏小鼠中测量的结果没有显着差异。在接受任一剂量的 PSI-7977 的对照小鼠或具有人源化肝脏的小鼠中,血浆乳酸水平也没有升高。
|
||
| 细胞实验 |
在 T75 烧瓶中,使用补充有 10% 胎牛血清、100 IU/mL 青霉素、和 100 μg/mL 链霉素。以类似的方式,使用细胞铺板培养基在每个烧瓶中接种约 5×106 人原代肝细胞。将细胞与 50 μM PSI-7851、PSI-7976 或 Sofosbuvir (PSI-7977) 在新鲜培养基中(对于克隆 A 细胞)或细胞维持培养基(对于原代肝细胞)在 37°C、5% CO2 中孵育长达 24 小时放置过夜后2 个大气压。在研究中使用放射性标记的 PSI-7851 时,遵循相同的方案,不同之处在于将 1×106 细胞接种到 6 孔板的每个孔中,然后将细胞孵育含有 5 μM [3H]PSI-7851。每隔预定的时间间隔取出培养基,并使用冷的磷酸盐缓冲盐水(PBS)清洗细胞层。胰蛋白酶消化后,计数细胞并以 1,200 rpm 离心 5 分钟。将细胞沉淀悬浮于 1 mL 冷的 60% 甲醇中后,在 -20°C 下放置过夜。将样品以 14,000 rpm 离心 5 分钟后,收集上清液,用 SpeedVac 浓缩器干燥,并保存在 -20°C 直至进行高效液相色谱 (HPLC) 分析。将残留物悬浮在 100 μL 水中后,将 50 μL 等份注入 HPLC。
|
||
| 动物实验 |
|
||
| 药代性质 (ADME/PK) |
Absorption, Distribution and Excretion
When given orally, sofosbuvir reaches its maximum plasma concentration in about 0.5 to 2 hours with a maximal concentration (Cmax) of 567 ng/mL. Sofosbuvir is eliminated by three routes: urine ( 80%), feces (14%), and respiration (2.5%); however, elimination through the kidneys is the major route. The volume of distribution for sofosbuvir has yet to be determined. The clearance of sofosbuvir has yet to be determined. Sofosbuvir is approximately 61-65% bound to human plasma proteins and the binding is independent of drug concentration over the range of 1 ug/mL to 20 ug/mL. Protein binding of GS-331007 was minimal in human plasma. After a single 400 mg dose of (14)C-sofosbuvir in healthy subjects, the blood to plasma ratio of (14)C-radioactivity was approximately 0.7. The pharmacokinetic properties of sofosbuvir and the predominant circulating metabolite GS-331007 have been evaluated in healthy adult subjects and in subjects with chronic hepatitis C. Following oral administration of SOVALDI, sofosbuvir was absorbed with a peak plasma concentration observed at approximately 0.5-2 hour post-dose, regardless of dose level. Peak plasma concentration of GS-331007 was observed between 2 to 4 hours post-dose. Based on population pharmacokinetic analysis in subjects with genotype 1 to 6 HCV infection who were coadministered ribavirin (with or without pegylated interferon), geometric mean steady state AUC0-24 was 969 ng*hr/mL for sofosbuvir (N=838), and 6790 ng*hr/mL for GS-331007 (N=1695), respectively. Relative to healthy subjects administered sofosbuvir alone (N = 272), the sofosbuvir AUC0-24 was 60% higher; and GS-331007 AUC0-24 was 39% lower, respectively, in HCV-infected subjects. Sofosbuvir and GS-331007 AUCs are near dose proportional over the dose range of 200 mg to 1200 mg. Following a single 400 mg oral dose of (14)C-sofosbuvir, mean total recovery of the dose was greater than 92%, consisting of approximately 80%, 14%, and 2.5% recovered in urine, feces, and expired air, respectively. The majority of the sofosbuvir dose recovered in urine was GS-331007 (78%) while 3.5% was recovered as sofosbuvir. These data indicate that renal clearance is the major elimination pathway for GS-331007. Studies in pregnant rats showed that sofosbuvir crossed the placenta. Fetal blood and brain sofosbuvir derived radioactivity was higher than in dams, but fetal liver and kidney had lower levels than corresponding organs in dams. Sofosbuvir-derived radioactivity was also quantifiable in milk from day 2 postpartum rats, but nursing pups did not appear to be extensively exposed to drug-derived radioactivity. Milk to plasma ratios were 0.1 at 1 hour and 0.8 at 24 hours. For more Absorption, Distribution and Excretion (Complete) data for Sofosbuvir (6 total), please visit the HSDB record page. Metabolism / Metabolites In vitro studies in human liver microsomes showed that sofosbuvir was an efficient substrate for Cathepsin A (Cat A) and carboxyl esterase 1 (CES1). Sofosbuvir was cleaved by CatA and CES1 and subsequent activation steps included amino acid removal by histidine triad nucleotide-binding protein 1 (HINT1) and phosphorylation by uridine monophosphate-cytidine monophosphate (UMP-CMP) kinase and nucleoside diphosphate (NDP) kinase. In vitro data indicated that Cat A preferentially hydrolysed sofosbuvir (the S-diastereomer) while CES1 did not exhibit stereoselectivity. In vitro studies in human liver microsomes showed that sofosbuvir was an efficient substrate for Cathepsin A (Cat A) and carboxyl esterase 1 (CES1). There were no indications of metabolism via urdine diphosphate glucuronosyltransferases (UGTs) or flavin-containing monooxygenase (FMO). Sofosbuvir was cleaved by CatA and CES1 and subsequent activation steps included amino acid removal by histidine triad nucleotide-binding protein 1 (HINT1) and phosphorylation by uridine monophosphate-cytidine monophosphate (UMP-CMP) kinase and nucleoside diphosphate (NDP) kinase. In vitro data indicated that Cat A preferentially hydrolysed sofosbuvir (the S-diastereomer) while CES1 did not exhibit stereoselectivity. This would be consistent with studies using GS-9851 showing a less efficient metabolism to the triphosphate in the hepatically-derived cell line containing the Clone A replicon and shown to exhibit low CES 1 activity, but high Cat A activity compared with primary human hepatocytes. Following incubation of hepatocytes from rat, dog, monkey and human GS-9851 was converted to the triphosphate GS-461203 in all species, most efficiently in human. Sofosbuvir was also readily converted to the triphosphate in dog liver after oral doses and was the dominant metabolite at all time points assessed with a long half-life of approx. 18 hours. The active metabolite GS-461203 could not be detected in monkey. Further while GS-461203 was detected in rat liver, it could not be measured in liver from mouse. Sofosbuvir is extensively metabolized in the liver to form the pharmacologically active nucleoside analog triphosphate GS-461203. The metabolic activation pathway involves sequential hydrolysis of the carboxyl ester moiety catalyzed by human cathepsin A (CatA) or carboxylesterase 1 (CES1) and phosphoramidate cleavage by histidine triad nucleotide-binding protein 1 (HINT1) followed by phosphorylation by the pyrimidine nucleotide biosynthesis pathway. Dephosphorylation results in the formation of nucleoside metabolite GS-331007 that cannot be efficiently rephosphorylated and lacks anti-HCV activity in vitro. GS-331007 and GS-566500 were detected in all species with GS-331007 being the major drug related material in all species and all matrices. In plasma, urine and feces of all species administered sofosbuvir the primary metabolite detected was GS-331007 accounting for >80% of total exposure. In rat liver and plasma GS-566500 was also detected. The metabolite profile was overall comparable between non-pregnant, pregnant and postpartum rats and in milk of postpartum rats with GS-331007 and 2 sulfate conjugates of GS-331007 being the major metabolites. In dog following a single oral dose of 20 mg/kg of sofosbuvir three metabolites in plasma were identified, GS-331007, GS-566500 and M4 (proposed glucuronidation product of GS-606965), accounting for 93.4%, 1.6% and 0.5%, respectively of total plasma AUC. Parent compound amounted to 4.5%. In dog (and mouse) the majority of a radioactive dose was recovered in urine within 8 to 12 hours. For more Metabolism/Metabolites (Complete) data for Sofosbuvir (7 total), please visit the HSDB record page. Biological Half-Life Sofosbuvir has a terminal half life of 0.4 hours. The median terminal half-lives of sofosbuvir and GS-331007 were 0.4 and 27 hours, respectively. |
||
| 毒性/毒理 (Toxicokinetics/TK) |
Toxicity Summary
IDENTIFICATION AND USE: Sofosbuvir is a white to off-white crystalline solid. Sofosbuvir is a direct-acting antiviral agent (pan-genotypic polymerase inhibitor) against the hepatitis C virus. It is used in conjunction with other antiviral agents for the treatment of chronic hepatitis C virus (HCV) genotype 1, 2, 3, or 4 infections in adults, including those with hepatocellular carcinoma awaiting liver transplantation and those with human immunodeficiency virus (HIV) co-infection. Sofosbuvir must be used as part of a multiple-drug regimen and should not be used alone for the treatment of chronic HCV infection. HUMAN EXPOSURE AND TOXICITY: The highest documented dose of sofosbuvir was a single supratherapeutic dose of sofosbuvir 1200 mg administered to 59 healthy subjects. There were no untoward effects observed at this dose level, and adverse events were similar in frequency and severity to those reported in the placebo and sofosbuvir 400 mg treatment groups. Sofosbuvir did not induce chromosome aberration using human peripheral blood lymphocytes. ANIMAL STUDIES: Single dose toxicity study was performed with GS-9851/PSI-7851 (the diastereomeric mixture) in rats. No mortality, clinical signs, body weight changes, macroscopic pathology, or organ weight changes for liver and kidney up to a highest dose of 1,800 mg/kg. Sofosbuvir or GS-9851, a 1:1 diastereomeric mixture of sofosbuvir and its stereoisomer, were evaluated in repeat-dose oral toxicity studies up to 13 weeks in mice, 26 weeks in rats, and 39 weeks in dogs. The primary target organs identified were the cardiovascular, hepatobiliary, gastrointestinal (GI) and hematopoietic (erythroid) systems. In the 7-day toxicity studies with GS-9851 doses of 2000 mg/kg/day in the rat and 1500 mg/kg/day in the dog resulted (but were not limited to) in increased mucus secretions in the stomach, glycogen depletion, and increased alanine aminotransferase (ALT), aspartate aminotransferase (AST), and bilirubin, with associated histopathologic liver findings in dogs; and heart adverse effects in rats (e.g., multifocal cardiac myofiber degeneration) and dogs (e.g., increased QT/QTc intervals). Findings in the liver and heart were not observed in long-term studies with GS-9851 or sofosbuvir. In chronic toxicity studies in rats (26 weeks) and dogs (39 weeks), effects included (but were not limited to) GI-related clinical signs (e.g., soft feces and emesis) and a decrease (e.g., approximately 10%) in mean red cell indices that were observed mainly in the high-dose group of dogs. Sofosbuvir had no effects on embryo-fetal viability or on fertility when evaluated in rats. No teratogenic effects were observed in rat and rabbit developmental toxicity studies with sofosbuvir. It had no adverse effects on behavior, reproduction, or development of the offspring in the rat pre- and post-natal development study. At the highest dose tested, exposure to the predominant circulating metabolite GS-331007 was at least 8-fold the exposure in humans at the recommended clinical dose. Fertility was normal in the offspring of rats exposed daily from before birth (in utero) through lactation day 20 at daily GS-331007 exposures (AUC) of approximately 12-fold higher than human exposures at the recommended clinical dose. Two-year carcinogenicity studies in mice and rats were conducted with sofosbuvir. Mice were administered doses of up to 200 mg/kg/day in males and 600 mg/kg/day in females, while rats were administered doses of up to 750 mg/kg/day in males and females. No increase in the incidence of drug-related neoplasms were observed at the highest doses tested in mice and rats, resulting in AUC exposure to the predominant circulating metabolite GS-331007 of approximately 7- and 30-fold (in mice) and 13- and 17-fold (in rats), in males and females respectively, the exposure in humans at the recommended clinical dose. Sofosbuvir was not genotoxic in a battery of in vitro or in vivo assays, including bacterial mutagenicity, and in vivo mouse micronucleus assays. Interactions Concomitant use of rifampin, a potent inducer of P-gp in the intestine, and sofosbuvir may cause decreased plasma concentrations of sofosbuvir and GS-331007 and may lead to decreased therapeutic effect of sofosbuvir. Rifampin and sofosbuvir should not be used concomitantly. Rifabutin is expected to cause decreased plasma concentrations of sofosbuvir and GS-331007, which may lead to decreased therapeutic effect of sofosbuvir. Concomitant use of rifabutin and sofosbuvir is not recommended. When used concomitantly with sofosbuvir, certain anticonvulsants (i.e., carbamazepine, oxcarbazepine, phenobarbital, phenytoin) are expected to decrease plasma concentrations of sofosbuvir and GS-331007, which may lead to decreased therapeutic effect of sofosbuvir. Concomitant use of these anticonvulsants and sofosbuvir is not recommended. Sofosbuvir is a substrate of breast cancer resistance protein (BCRP); GS-331007 is not a BCRP substrate. Inhibitors of BCRP may cause increased plasma concentrations of sofosbuvir without increasing plasma concentrations of GS-331007. Sofosbuvir and GS-331007 are not BCRP inhibitors; pharmacokinetic interactions are unlikely with drugs that are BCRP substrates. For more Interactions (Complete) data for Sofosbuvir (13 total), please visit the HSDB record page. |
||
| 参考文献 |
|
||
| 其他信息 |
Therapeutic Uses
Sovaldi is a hepatitis C virus (HCV) nucleotide analog NS5B polymerase inhibitor indicated for the treatment of chronic hepatitis C (CHC) infection as a component of a combination antiviral treatment regimen. /Included in US product label/ The following points should be considered when initiating treatment with Sovaldi: Monotherapy of Sovaldi is not recommended for treatment of chronic hepatitis C (CHC). Treatment regimen and duration are dependent on both viral genotype and patient population. Treatment response varies based on baseline host and viral factors. Drug Warnings FDA is warning that serious slowing of the heart rate can occur when the antiarrhythmic drug amiodarone is taken together with either the hepatitis C drug Harvoni (ledipasvir/sofosbuvir) or with Sovaldi (sofosbuvir) taken in combination with another direct acting antiviral for the treatment of hepatitis C infection. FDA is adding information about serious slowing of the heart rate, known as symptomatic bradycardia, to the Harvoni and Sovaldi labels. FDA is recommending that health care professionals should not prescribe either Harvoni or Sovaldi combined with another direct acting antiviral, such as the investigational drug daclatasvir or Olysio (simeprevir), with amiodarone. FDA review of submitted postmarketing adverse event reports found that patients can develop a serious and life-threatening symptomatic bradycardia when either Harvoni or Sovaldi combined with another direct-acting antiviral is taken together with amiodarone. The reports included the death of one patient due to cardiac arrest and three patients requiring placement of a pacemaker to regulate their heart rhythms. The other patients recovered after discontinuing either the hepatitis C drugs or amiodarone, or both. The cause of these events could not be determined. FDA will continue to monitor Harvoni and Sovaldi for risks of serious symptomatic bradycardia and further investigate the reason why the use of amiodarone with these hepatitis C drugs led to the heart-related events. Concomitant use of sofosbuvir with drugs that are potent inducers of the P-glycoprotein (P-gp) transport system in the intestine (e.g., rifampin, St. John's wort) is not recommended since this may result in substantially decreased sofosbuvir plasma concentrations and could lead to reduced therapeutic effect of sofosbuvir. Anemia has been reported in patients receiving sofosbuvir in conjunction with ribavirin or in conjunction with peginterferon alfa andribavirin. In clinical trials, anemia was reported in 21% of patients who received 12 weeks of treatment with sofosbuvir, peginterferon alfa, and ribavirin compared with 12% of patients who received 24 weeks of treatment with peginterferon alfa and ribavirin without sofosbuvir. In addition, hemoglobin concentrations less than 10 g/dL were reported in 23% of patients who received 12 weeks of treatment with sofosbuvir, peginterferon alfa, and ribavirin compared with 14% of patients who received 24 weeks of treatment with peginterferon alfa and ribavirin without sofosbuvir. Adverse effects reported in more than 20% of patients receiving sofosbuvir in conjunction with ribavirin and peginterferon alfa include fatigue, headache, nausea, insomnia, and anemia. For more Drug Warnings (Complete) data for Sofosbuvir (13 total), please visit the HSDB record page. Pharmacodynamics Sofosbuvir acts against HCV and is categorized as a direct-acting antiviral agent (DAA). At a dose 3 times the recommended dose, sofosbuvir does not prolong QTc to any clinically relevant extent. |
| 分子式 |
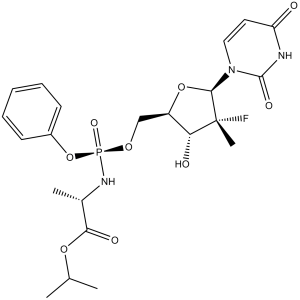
C22H29FN3O9P
|
|
|---|---|---|
| 分子量 |
529.45
|
|
| 精确质量 |
529.162
|
|
| 元素分析 |
C, 49.91; H, 5.52; F, 3.59; N, 7.94; O, 27.20; P, 5.85
|
|
| CAS号 |
1190307-88-0
|
|
| 相关CAS号 |
|
|
| PubChem CID |
45375808
|
|
| 外观&性状 |
White solid powder
|
|
| 密度 |
1.4±0.1 g/cm3
|
|
| 折射率 |
1.573
|
|
| LogP |
1.62
|
|
| tPSA |
167.99
|
|
| 氢键供体(HBD)数目 |
3
|
|
| 氢键受体(HBA)数目 |
11
|
|
| 可旋转键数目(RBC) |
11
|
|
| 重原子数目 |
36
|
|
| 分子复杂度/Complexity |
913
|
|
| 定义原子立体中心数目 |
6
|
|
| SMILES |
O=C1N([C@H]2[C@]([C@H](O)[C@@H](CO[P@](OC3=CC=CC=C3)(N[C@@H](C)C(OC(C)C)=O)=O)O2)(C)F)C=CC(N1)=O
|
|
| InChi Key |
TTZHDVOVKQGIBA-IQWMDFIBSA-N
|
|
| InChi Code |
InChI=1S/C22H29FN3O9P/c1-13(2)33-19(29)14(3)25-36(31,35-15-8-6-5-7-9-15)32-12-16-18(28)22(4,23)20(34-16)26-11-10-17(27)24-21(26)30/h5-11,13-14,16,18,20,28H,12H2,1-4H3,(H,25,31)(H,24,27,30)/t14-,16+,18+,20+,22+,36-/m0/s1
|
|
| 化学名 |
propan-2-yl (2S)-2-[[[(2R,3R,4R,5R)-5-(2,4-dioxopyrimidin-1-yl)-4-fluoro-3-hydroxy-4-methyloxolan-2-yl]methoxy-phenoxyphosphoryl]amino]propanoate
|
|
| 别名 |
|
|
| HS Tariff Code |
2934.99.9001
|
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
|
|||
|---|---|---|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 1.67 mg/mL (3.15 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 16.7 mg/mL澄清的DMSO储备液加入到400 μL PEG300中,混匀;再向上述溶液中加入50 μL Tween-80,混匀;然后加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: ≥ 1.67 mg/mL (3.15 mM) (饱和度未知) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 16.7mg/mL澄清的DMSO储备液加入到900μL 20%SBE-β-CD生理盐水中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 View More
配方 3 中的溶解度: ≥ 1.67 mg/mL (3.15 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 配方 4 中的溶解度: 4.55 mg/mL (8.59 mM) in PBS (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液; 超声助溶. 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.8888 mL | 9.4438 mL | 18.8875 mL | |
| 5 mM | 0.3778 mL | 1.8888 mL | 3.7775 mL | |
| 10 mM | 0.1889 mL | 0.9444 mL | 1.8888 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
| NCT Number | Recruitment | interventions | Conditions | Sponsor/Collaborators | Start Date | Phases |
| NCT03855917 | Recruiting | Drug: Sofosbuvir 400mg [Sovaldi] Drug: Glecaprevir/pibrentasvir (300mg/120mg) |
Hepatitis C | Kirby Institute | February 11, 2020 | Phase 4 |
| NCT03540212 | Recruiting | Drug: Daclatasvir and sofosbuvir | Chronic HCV Infection | Ain Shams University | January 2009 | Phase 2 Phase 3 |
| NCT03646396 | Recruiting | Drug: Sofosbuvir-daclatasvir | HCV Coinfection | Sherief Abd-Elsalam | August 1, 2018 | Not Applicable |
| NCT05854511 | Recruiting | Drug: Sofosbuvir 200 MG Oral Tablet plus Daclatasvir 30 mg Oral tablets |
HCV | Ain Shams University | June 5, 2022 | Phase 3 |
| NCT04852614 | Recruiting | Diagnostic Test: Pharmacokinetic test |
Hepatitis C Virus Infection | Ain Shams University | December 1, 2020 | N/A |
|
 RNase L-IN-2
RNase L-IN-2
 BW 348U87
BW 348U87
 ERCC1/XPA interaction inhibitor 1
ERCC1/XPA interaction inhibitor 1
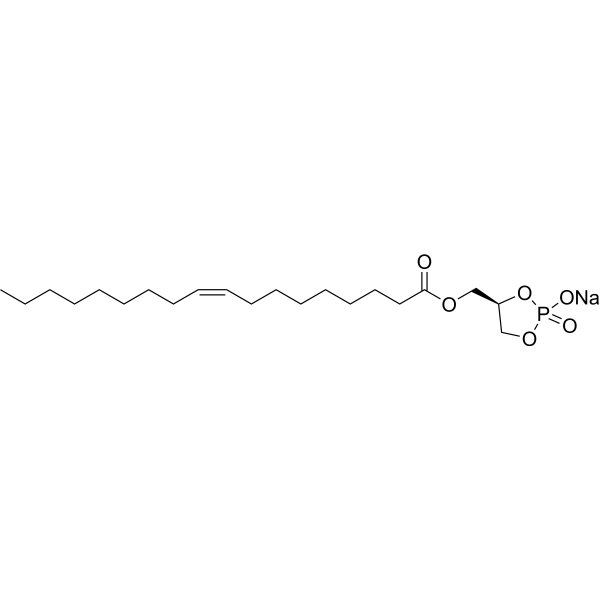 PHYLPA-8
PHYLPA-8
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1





























 COA
COA

