
| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 1g |
|
||
| 2g |
|
||
| 5g |
|
||
| 10g |
|
||
| 25g |
|
||
| Other Sizes |
|

| 靶点 |
DPP-4 (IC50 = 18 nM)
|
|---|---|
| 体外研究 (In Vitro) |
Sitagliptin 磷酸盐可有效抑制 Caco-2 细胞提取物中的 DPP-4,IC50 为 19 nM [1]。西他列汀通过激活 cAMP/PKA/Rac1 通路抑制体外分离的脾 CD4 T 细胞的迁移 [2]。根据最近的一项研究,西格列汀通过独特的直接作用刺激肠道 L 细胞产生 GLP-1,该作用依赖于 MEK-ERK1/2 和蛋白激酶 A,但不依赖于 DPP-4。因此,它减轻了自身免疫对移植物存活的负面影响[3]。
|
| 体内研究 (In Vivo) |
在随意喂养的 Han-Wistar 大鼠体内,磷酸西格列汀抑制血浆 DPP-4 活性的 ED50 测定为给药后 7 小时 2.3 mg/kg 和给药后 24 小时 30 mg/kg [1]。链脲佐菌素引起的 1 型糖尿病小鼠模型血浆 DPP-4 水平升高;给予高磷酸西他列汀饮食的小鼠,这种蛋白质的水平要低得多。这是通过改善高血糖的管理来实现的,也许可以通过延长胰岛移植的存活率来实现[4]。大鼠磷酸西他列汀的分布容积和血浆清除率(40-48 mL/min/kg,7-9 L/kg)比狗(9 mL/min/kg,3 L/kg)更大;该药物在大鼠中的半衰期(2 小时)比在狗中的半衰期(4 小时)短[5]。
|
| 酶活实验 |
汇合的 Caco-2 细胞用于提取 DPP-4。用裂解缓冲液(10 mM Tris-HCl、150 mM NaCl、0.04 U/mL 抑肽酶、0.5% Nonidet P40、pH 8.0)在室温下孵育 5 分钟后,将细胞在 35,000 g、4 ℃下离心 30 分钟。 °C,然后将上清液保存在-80°C。将二十微升合适的化合物稀释液与五十微升作为 DPP-4 酶底物的 H-Ala-Pro-7-酰胺基-4-三氟甲基香豆素(测定中的最终浓度:100 微升)和三十微升Caco-2 细胞提取物(用 100 mM Tris-HCl、100 mM NaCl、pH 7.8 稀释 1000 倍)。将板在室温下孵育一小时后,使用 SpectraMax GeminiXS 在 405/535 nm 的激发/发射波长下测量荧光。将 Caco-2 细胞提取物暴露于高浓度抑制剂(BI 1356 为 30 nM,维格列汀为 3 μM)一小时后,确定抑制剂与 DPP-4 酶的解离动力学。一旦用测定缓冲液将预孵育混合物稀释 3000 倍,就通过添加底物 H-Ala-Pro-7-amido-4-triflumethylcoumarini 来启动酶促反应。仍然与DPP-4酶结合的抑制剂的量通过在存在或不存在抑制剂的情况下给定时间的DPP-4活性的差异来指示。使用 SpectraMax 的 SoftMax 软件,以 10 分钟的间隔计算最大反应速率(荧光单位/秒 × 1000),并针对未抑制反应的速率进行校正 [(vcontrol-vinhibitor)/vcontrol]。
|
| 细胞实验 |
将含有 CD4T 细胞的膜插入物铺板于无血清 RPMI 1640 中。使用 Corning Transwell 小室测量细胞迁移,使用或不使用 DPP-4 抑制剂 (100 μM) 和纯化的猪肾 DPP-4(32.1 单位/毫克;最终浓度为 100 mU/mL)。一小时后,对移入下室的细胞进行计数,并机械去除上表面的细胞。迁移量的表达式与对照样品相关。
胰高血糖素样肽-1(GLP-1)是由肠L细胞分泌到循环中的肠促胰岛素激素。二肽基肽酶IV(DPP-IV)抑制剂西格列汀可防止GLP-1降解,并在临床上用于治疗2型糖尿病患者,从而改善糖化血红蛋白水平。当在2型糖尿病模型新生链脲佐菌素大鼠中检查西格列汀对GLP-1水平的影响时,观察到活性GLP-1的基础血浆水平增加4.9±0.9倍,口服葡萄糖刺激的血浆水平增加3.6±0.4倍(P<0.001),肠道L细胞总数增加1.5±0.1倍(P<0.01)。因此,在体外小鼠GLUTag(mGLUTag)和人hNCI-H716肠L细胞中研究了西格列汀对GLP-1分泌和L细胞信号传导的直接影响。西格列汀(0.1-2μM)增加了mGLUTag和hNCI-H716细胞的GLP-1总分泌量(P<0.01-0.001)。然而,MK0626(1-50μM)是一种结构上无关的DPP-IV抑制剂,在两种模型中均不影响GLP-1的分泌。用GLP-1受体激动剂毒蜥外泌肽-4处理mGLUTag细胞没有调节GLP-1的释放,表明GLP-1对L细胞没有反馈作用。西格列汀增加了mGLUTag和hNCI-H716细胞中的cAMP水平(P<0.01)和ERK1/2磷酸化(P<0.05),但没有改变细胞内钙或磷酸化Akt水平。用蛋白激酶A(H89和蛋白激酶抑制剂)或MAPK激酶-ERK1/2(PD98059和U0126)抑制剂预处理mGLUTag细胞可防止西格列汀诱导的GLP-1分泌(P<0.05-0.01)。这些研究首次证明,西格列汀对肠道L细胞具有直接的、不依赖DPP IV的作用,激活cAMP和ERK1/2信号传导,刺激GLP-1的总分泌[3]。 |
| 动物实验 |
Mice: C57BL/6J mice that have been fasted overnight are challenged with an oral glucose load (2 g/kg) 45 minutes after the compound is administered. Tail bleed predose and successive time points following the glucose load are used to draw blood samples for glucose measurement. DPP-4 inhibitors or a vehicle are given 16 hours prior to the glucose challenge in order to assess how long the effect lasts on glucose tolerance.
Effects of MK0431 on islet graft survival in diabetic NOD mice were determined with metabolic studies and micropositron emission tomography imaging, and its underlying molecular mechanisms were assessed. Results: Treatment of NOD mice with MK0431 before and after islet transplantation resulted in prolongation of islet graft survival, whereas treatment after transplantation alone resulted in small beneficial effects compared with nontreated controls. Subsequent studies demonstrated that MK0431 pretreatment resulted in decreased insulitis in diabetic NOD mice and reduced in vitro migration of isolated splenic CD4+ T-cells. Furthermore, in vitro treatment of splenic CD4+ T-cells with DPP-IV resulted in increased migration and activation of protein kinase A (PKA) and Rac1. Conclusions: Treatment with MK0431 therefore reduced the effect of autoimmunity on graft survival partially by decreasing the homing of CD4+ T-cells into pancreatic beta-cells through a pathway involving cAMP/PKA/Rac1 activation.[2] Effects of the DPP-IV inhibitor MK0431 (sitagliptin) on glycemic control and functional islet mass in a streptozotocin (STZ)-induced type 1 diabetes mouse model were determined with metabolic studies and microPET imaging. Results: The type 1 diabetes mouse model exhibited elevated plasma DPP-IV levels that were substantially inhibited in mice on an MK0431 diet. Residual beta-cell mass was extremely low in STZ-induced diabetic mice, and although active GLP-1 levels were increased by the MK0431 diet, there were no significant effects on glycemic control. After islet transplantation, mice fed normal diet rapidly lost their ability to regulate blood glucose, reflecting the suboptimal islet transplant. By contrast, the MK0431 group fully regulated blood glucose throughout the study, and PET imaging demonstrated a profound protective effect of MK0431 on islet graft size. Conclusions: Treatment with a DPP-IV inhibitor can prolong islet graft retention in an animal model of type 1 diabetes.[4] The pharmacokinetics, metabolism, and excretion of sitagliptin [MK-0431; (2R)-4-oxo-4-[3-(trifluoromethyl)-5,6-dihydro[1,2,4]triazolo[4,3-a]pyrazin-7(8H)-yl]-1-(2,4,5-trifluorophenyl)butan-2-amine], a potent dipeptidyl peptidase 4 inhibitor, were evaluated in male Sprague-Dawley rats and beagle dogs. The plasma clearance and volume of distribution of sitagliptin were higher in rats (40-48 ml/min/kg, 7-9 l/kg) than in dogs ( approximately 9 ml/min/kg, approximately 3 l/kg), and its half-life was shorter in rats, approximately 2 h compared with approximately 4 h in dogs. Sitagliptin was absorbed rapidly after oral administration of a solution of the phosphate salt. The absolute oral bioavailability was high, and the pharmacokinetics were fairly dose-proportional. After administration of [(14)C]sitagliptin, parent drug was the major radioactive component in rat and dog plasma, urine, bile, and feces. Sitagliptin was eliminated primarily by renal excretion of parent drug; biliary excretion was an important pathway in rats, whereas metabolism was minimal in both species in vitro and in vivo. Approximately 10 to 16% of the radiolabeled dose was recovered in the rat and dog excreta as phase I and II metabolites, which were formed by N-sulfation, N-carbamoyl glucuronidation, hydroxylation of the triazolopiperazine ring, and oxidative desaturation of the piperazine ring followed by cyclization via the primary amine. The renal clearance of unbound drug in rats, 32 to 39 ml/min/kg, far exceeded the glomerular filtration rate, indicative of active renal elimination of parent drug.[5] |
| 药代性质 (ADME/PK) |
Absorption
Sitagliptin is 87% orally bioavailable and taking it with or without food does not affect its pharmacokinetics. Sitagliptin reaches maximum plasma concentration in 2 hours. Route of Elimination Approximately 79% of sitagliptin is excreted in the urine as the unchanged parent compound. 87% of the dose is eliminated in the urine and 13% in the feces. Volume of Distribution 198L. Clearance 350mL/min. Sitagliptin is secreted in the milk of lactating rats at a milk to plasma ratio of 4:1. It is not known whether sitagliptin is excreted in human milk. Placental transfer of sitagliptin administered to pregnant rats was approximately 45% at 2 hours and 80% at 24 hours postdose. Placental transfer of sitagliptin administered to pregnant rabbits was approximately 66% at 2 hours and 30% at 24 hours. Approximately 79% of sitagliptin is excreted unchanged in the urine with metabolism being a minor pathway of elimination. Elimination of sitagliptin occurs primarily via renal excretion and involves active tubular secretion. Sitagliptin is a substrate for human organic anion transporter-3 (hOAT-3), which may be involved in the renal elimination of sitagliptin. The clinical relevance of hOAT-3 in sitagliptin transport has not been established. Sitagliptin is also a substrate of p-glycoprotein, which may also be involved in mediating the renal elimination of sitagliptin. However, cyclosporine, a p-glycoprotein inhibitor, did not reduce the renal clearance of sitagliptin. View More
Metabolism / Metabolites
Biological Half-Life Approximately 12.4 hours. Other studies have reported a half life of approximately 11 hours. Two double-blind, randomized, placebo-controlled, alternating-panel studies evaluated the safety, tolerability, pharmacokinetics, and pharmacodynamics of single oral doses of sitagliptin (1.5-600 mg) in healthy male volunteers. Sitagliptin was well absorbed (approximately 80% excreted unchanged in the urine) with an apparent terminal half-life ranging from 8 to 14 hours. ... PMID:16338283 The apparent terminal half life following a 100 mg oral dose of sitagliptin was approximately 12.4 hours |
| 毒性/毒理 (Toxicokinetics/TK) |
Toxicity Summary
IDENTIFICATION AND USE: Sitagliptin is a viscous liquid. It is a dipeptidyl peptidase-4 inhibitor and used to improve glycemic control in patients with type 2 diabetes. HUMAN EXPOSURE AND TOXICITY: Sitagliptin improves glycemic control and is generally well-tolerated in patients with type 2 diabetes. Sitagliptin use has been associated with an increased risk of heart failure -related hospitalizations among patients with type 2 diabetes with pre-existing heart failure. More recently a study has pointed to the possible use of sitagliptin in the treatment of some neurodegenerative conditions of the peripheral nervous system. Sitagliptin appears to be free from the adverse effects of weight gain and hypoglycemia that are associated some other treatments. ANIMAL STUDIES: Renal and liver toxicity were observed in rodents at systemic exposure to sitagliptin at values 58 times the human exposure level. Transient treatment-related physical signs, some of which suggest neural toxicity, such as open-mouth breathing, salivation, white foamy emesis, ataxia, trembling, decreased activity, and/or hunched posture were observed in dogs at exposure levels approximately 23 times the clinical exposure level. Carcinogenicity studies in mice did not show an increased incidence of tumors in any organ up to 500 mg/kg, but in rats there was an increased incidence of combined liver adenoma/carcinoma in males and females and of liver carcinoma in females at 500 mg/kg. Reproductive effects in rats and rabbits were only seen at doses greater than 250 mg/kg. Incisor teeth abnormalities were observed in rats at exposure levels 67 times the clinical exposure level. Sitagliptin was not mutagenic or clastogenic with or without metabolic activation in the Ames bacterial mutagenicity assay, a Chinese hamster ovary (CHO) chromosome aberration assay, an in vitro cytogenetics assay in CHO cells, an in vitro rat hepatocyte DNA alkaline elution assay, and an in vivo micronucleus assay. Hepatotoxicity Liver injury due to sitagliptin is rare. In large clinical trials, serum enzyme elevations were no more common with sitagliptin therapy (0.5%) than with placebo (0.4%), and no instances of clinically apparent liver injury were reported. Since licensure, instances of serum enzyme elevations attributed to sitagliptin have been reported to the FDA and the sponsor. A single case report of clinically apparent liver injury has been published, but in a patient who also had hepatitis C. The pattern of serum enzyme elevations was hepatocellular and peak serum bilirubin was 9.4 mg/dL, with a rapid recovery upon stopping sitagliptin. Immunoallergic features and autoantibodies were absent. Likelihood score: D (possible rare cause of clinically apparent liver injury). View More
Effects During Pregnancy and Lactation
Non-Human Toxicity Values LD50 Mouse oral 4000 mg/kg LD50 Rat oral >3000 mg/kg Protein Binding 38%. |
| 参考文献 |
|
| 其他信息 |
Sitagliptin Phosphate is the phosphate salt form of sitagliptin, an orally available, competitive, beta-amino acid-derived inhibitor of dipeptidyl peptidase 4 (DDP-4) with hypoglycemic activity. Sitagliptin may cause an increased risk in the development of pancreatitis.
A pyrazine-derived DIPEPTIDYL-PEPTIDASE IV INHIBITOR and HYPOGLYCEMIC AGENT that increases the levels of the INCRETIN hormones GLUCAGON-LIKE PEPTIDE-1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP). It is used in the treatment of TYPE 2 DIABETES. See also: Sitagliptin (has active moiety); Ertugliflozin; Sitagliptin Phosphate (component of); Metformin Hydrochloride; Sitagliptin Phosphate (component of) ... View More ... |
| 分子式 |
C₁₆H₁₈F₆N₅O₅P
|
|---|---|
| 分子量 |
505.31
|
| 精确质量 |
505.094
|
| 元素分析 |
C, 38.03; H, 3.59; F, 22.56; N, 13.86; O, 15.83; P, 6.13
|
| CAS号 |
654671-78-0
|
| 相关CAS号 |
Sitagliptin;486460-32-6;Sitagliptin-d4 phosphate;1432063-88-1;Sitagliptin phosphate monohydrate;654671-77-9;(S)-Sitagliptin phosphate;823817-58-9;(Rac)-Sitagliptin;823817-56-7
|
| PubChem CID |
6451150
|
| 外观&性状 |
White to off-white solid powder
|
| 沸点 |
529.9ºC at 760 mmHg
|
| 熔点 |
202-204ºC
|
| 闪点 |
274.3ºC
|
| LogP |
1.726
|
| tPSA |
164.61
|
| 氢键供体(HBD)数目 |
4
|
| 氢键受体(HBA)数目 |
14
|
| 可旋转键数目(RBC) |
4
|
| 重原子数目 |
33
|
| 分子复杂度/Complexity |
616
|
| 定义原子立体中心数目 |
1
|
| SMILES |
C1CN2C(=NN=C2C(F)(F)F)CN1C(=O)C[C@@H](CC3=CC(=C(C=C3F)F)F)N.OP(=O)(O)O
|
| InChi Key |
IQFYVLUXQXSJJN-SBSPUUFOSA-N
|
| InChi Code |
InChI=1S/C16H15F6N5O.H3O4P/c17-10-6-12(19)11(18)4-8(10)3-9(23)5-14(28)26-1-2-27-13(7-26)24-25-15(27)16(20,21)22;1-5(2,3)4/h4,6,9H,1-3,5,7,23H2;(H3,1,2,3,4)/t9-;/m1./s1
|
| 化学名 |
(3R)-3-amino-1-[3-(trifluoromethyl)-6,8-dihydro-5H-[1,2,4]triazolo[4,3-a]pyrazin-7-yl]-4-(2,4,5-trifluorophenyl)butan-1-one;phosphoric acid
|
| 别名 |
Januvia; MK0431 phosphate; Sitagliptin phosphate; 654671-78-0; Sitagliptin (phosphate); MK-0431; sitagliptin phosphate anhydrous; UNII-494P4635I6; Sitagliptin monophosphate; MK 431; MK 0431 phosphate; MK-0431 phosphate
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month 注意: 请将本产品存放在密封且受保护的环境中(例如氮气保护),避免吸湿/受潮和光照。 |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
DMSO : ~100 mg/mL (~197.90 mM)
H2O : ~50 mg/mL (~98.95 mM) |
|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 2.5 mg/mL (4.95 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 25.0 mg/mL澄清DMSO储备液加入到400 μL PEG300中,混匀;然后向上述溶液中加入50 μL Tween-80,混匀;加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: ≥ 2.5 mg/mL (4.95 mM) (饱和度未知) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 25.0 mg/mL澄清DMSO储备液加入900 μL 20% SBE-β-CD生理盐水溶液中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 View More
配方 3 中的溶解度: ≥ 2.5 mg/mL (4.95 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 配方 4 中的溶解度: 50 mg/mL (98.95 mM) in PBS (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液; 超声助溶. 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.9790 mL | 9.8949 mL | 19.7898 mL | |
| 5 mM | 0.3958 mL | 1.9790 mL | 3.9580 mL | |
| 10 mM | 0.1979 mL | 0.9895 mL | 1.9790 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
| NCT Number | Recruitment | interventions | Conditions | Sponsor/Collaborators | Start Date | Phases |
| NCT05972928 | Not yet recruiting | Drug: Sitagliptin 100mg | Polycystic Ovary Syndrome | Beni-Suef University | July 30, 2023 | Phase 2 Phase 3 |
| NCT04495881 | Recruiting | Drug: Sitagliptin 100mg | Type 2 Diabetes | Beijing Chao Yang Hospital | January 1, 2020 | Phase 4 |
| NCT05219409 | Not yet recruiting | Drug: Sitagliptin Device: Professional CGM |
Type 1 Diabetes | University of Milan | July 2023 | Phase 2 Phase 3 |
| NCT04298684 | Not yet recruiting | Drug: Sitagliptin Drug: METFORMIN |
Diabetes Mellitus, Type 2 Thyroid Nodule (Benign) |
Centre Hospitalier Universitaire de Pointe-a-Pitre |
January 1, 2021 | Phase 4 |
| NCT05353673 | Recruiting | Drug: Sitagliptin Drug: Danazol |
Thrombocytopenia Immune Thrombocytopenia |
Peking University People's Hospital |
June 1, 2021 | Phase 2 |
 DPP-4-IN-11
DPP-4-IN-11
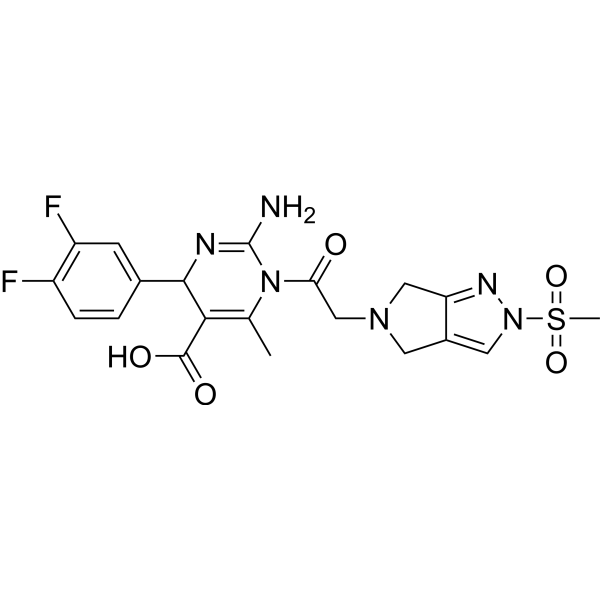 DPP-4-IN-12
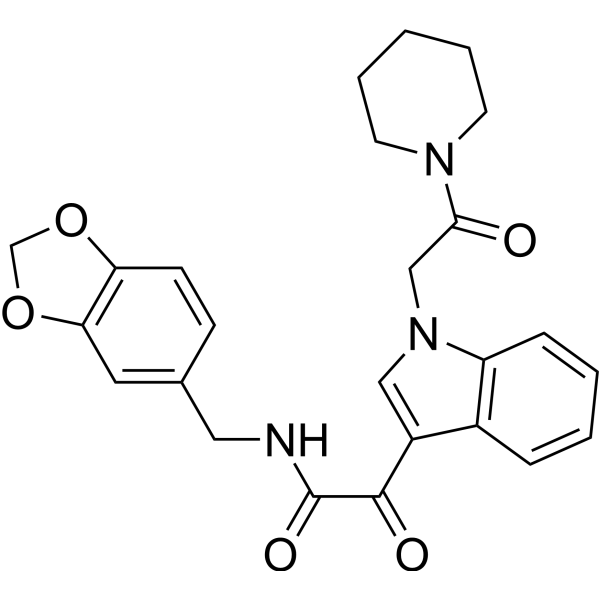
DPP-4-IN-12
 ZINC000003015356
ZINC000003015356
 SGP8
SGP8
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1
























