
| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 5mg |
|
||
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| 500mg |
|
||
| Other Sizes |
|

| 体外研究 (In Vitro) |
所有测试的单突变体和双突变体 (EC50=0.1-2.0 nM) 和野生型 HIV-1 (EC50=0.4 nM) 均对 R278474 的活性敏感[1]。 30 天内,R278474(10-5000 nM;30 天)在 1 μM 浓度下未显示任何野生型 HIV-1 突破的迹象[1]。在低于 1 nM 的 50% 抑制浓度 (EC50) 下,R278474 可抑制 81% 的临床分离株(约 1200 种重组临床分离株),在 EC50 低于 10 nM 时,可抑制 94%[1]。 TMC278 在 MT4 T 细胞中对 O 组分离株表现出纳摩尔 EC50 (2.88-8.45 nM),对野生型 HIV-1 M 组分离株表现出亚纳摩尔 EC50 (0.07-1.01 nM)[2]。
|
||
|---|---|---|---|
| 体内研究 (In Vivo) |
用 R278474(10-160 mg/kg;口服 1 个月)治疗的大鼠没有表现出任何异常效应,但较高剂量水平下肝脏重量增加和物种特异性甲状腺肥大除外[1]。在大鼠中,R278474 (iv) 的消除半衰期为 4.4 小时至 31 小时。在狗中,暴露量 (AUCinf) 为每公斤 8.7 小时 (1.25 mg/kg),在兔中为每公斤 1.4 小时 (1.25 mg/kg),以及每公斤 44 小时 (1.25 mg/kg)[1]。在大鼠和狗中,R278474 (po) 的半衰期分别为 2.8 和 39 小时,口服生物利用度分别为 32% 和 31%[1]。
|
||
| 动物实验 |
|
||
| 药代性质 (ADME/PK) |
Absorption, Distribution and Excretion
Rilpivirine has a Tmax of 3-4 hours and has a mean AUC of 2235 ± 851 ng\*h/mL. A 25mg dose reaches a Cmax of 247 ng/mL in healthy subjects and 138.6 ng/mL in patients with HIV-1. Rilpivirine is 85% eliminated in the feces and 6.1% eliminated in the urine. 25% of a dose is recovered in the feces as the unchanged parent drug, while <1% of a dose is recovered in the urine as the unchanged parent drug. In HIV-1 patients, the apparent volume of distribution in the central compartment was 152-173 L. In HIV-1 patients, the apparent total clearance is estimated to be 6.89-8.66 L/h. After a single oral dose, an average of 85% of the dose is eliminated in feces (75% as metabolites) and 6% is eliminated in urine (only trace amounts as unchanged rilpivirine). It is not known whether rilpivirine is distributed into human milk; however, the drug is distributed into milk in rats. Metabolism / Metabolites Rilpivirine is predominantly metabolized by CYP3A4 and CYP3A5 to the hydroxylated metabolites M1, M2, M3, and M4. UGT1A1 glucuronidates the M2 metabolite to form M6, UGT1A4 glucuronidates rilpivirine to form M5, and an unknown UGT glucuronidates the M4 metabolite to form M7. Rilpivirine is metabolized by the cytochrome P-450 (CYP) isoenzyme 3A. Biological Half-Life Rilpivirine has a terminal half-life of 34-55 hours. The terminal elimination half-life of rilpivirine is approximately 50 hours. |
||
| 毒性/毒理 (Toxicokinetics/TK) |
Hepatotoxicity
Serum aminotransferase elevations occur in 25% or more of patients on rilpivirine therapy, but elevations above 5 times the upper limit of normal are uncommon, occurring in 1% to 4% of patients. The rate of serum aminotransferase elevations during rilpivirine therapy is higher in patients who are coinfected with hepatitis B or C [~10% have values greater than 5 times ULN]. The product label for rilpivirine induces a warning about hepatotoxicity particularly in patients with HBV or HCV coinfection and recommends monitoring for liver test abnormalities. During the first several years of wide spread clinical use of rilpivirine, a single case report of liver injury was published. The case was marked by prominent elevations in serum ALT and AST without jaundice arising within days of starting therapy and resolving rapidly upon stopping (Case 1). There was no rash, eosinophilia or other prominent immunoallergic features which are typical of the liver injury associated with nevirapine and efavirenz. Thus, clinically apparent hepatotoxicity due to rilpivirine may occur but is rare. Liklihood score: D (possible rare cause of clinically apparent liver injury). Effects During Pregnancy and Lactation ◉ Summary of Use during Lactation Limited information indicates that maternal rilpivirine doses of 25 mg daily produce low levels in milk and infant serum. Until more data become available, an alternate drug may be preferred, especially while nursing a newborn or preterm infant. Achieving and maintaining viral suppression with antiretroviral therapy decreases breastfeeding transmission risk to less than 1%, but not zero. Individuals with HIV who are on antiretroviral therapy with a sustained undetectable viral load and who choose to breastfeed should be supported in this decision. If a viral load is not suppressed, banked pasteurized donor milk or formula is recommended. ◉ Effects in Breastfed Infants Relevant published information was not found as of the revision date. ◉ Effects on Lactation and Breastmilk Relevant published information was not found as of the revision date. Protein Binding Rilpivirine is >99% bound to plasma protein, most commonly albumin. Interactions Concomitant use of omeprazole and rilpivirine has resulted in decreased rilpivirine plasma concentrations and AUC. Concomitant use of other proton-pump inhibitors (e.g., esomeprazole, lansoprazole, pantoprazole, rabeprazole) also may result in decreased rilpivirine plasma concentrations. Concomitant use of rilpivirine and proton-pump inhibitors is contraindicated. Concomitant use of famotidine and rilpivirine has resulted in decreased rilpivirine plasma concentrations and area under the concentration-time curve (AUC). Concomitant use of other histamine H2-receptor antagonists (e.g., cimetidine, nizatidine, ranitidine) also may result in decreased rilpivirine plasma concentrations. Rilpivirine and histamine H2-receptor antagonists should be used concomitantly with caution; histamine H2-receptor antagonists should be administered at least 12 hours before or at least 4 hours after rilpivirine. Potential pharmacokinetic interaction with antacids such as aluminum hydroxide, calcium carbonate, or magnesium hydroxide (decreased plasma rilpivirine concentrations). Antacids and rilpivirine should be used concomitantly with caution; antacids should be administered at least 2 hours before or at least 4 hours after rilpivirine. Rilpivirine is metabolized by the cytochrome P-450 (CYP) isoenzyme 3A. Concomitant use with drugs that induce CYP3A may result in decreased plasma rilpivirine concentrations and may result in possible loss of virologic response and development of resistance to rilpivirine or the nonnucleoside reverse transcriptase inhibitor (NNRTI) class. Concomitant use with drugs that inhibit CYP3A may result in increased plasma rilpivirine concentrations. When the recommended rilpivirine dosage (25 mg once daily) is used, it is unlikely to have clinically important effects on the pharmacokinetics of drugs that are metabolized by CYP isoenzymes. |
||
| 参考文献 |
|
||
| 其他信息 |
Therapeutic Uses
HIV Reverse Transcriptase/antagonists & inhibitors Due to ongoing neuropsychiatric adverse events in some efavirenz (EFV)-treated patients, a switch to an alternative non-nucleoside reverse transcriptase inhibitor may be considered. Rilpivirine (RPV) has been coformulated as a single-tablet regimen (STR) with emtricitabine/tenofovir disoproxil fumarate (FTC/TDF), and the components have demonstrated noninferior efficacy to EFV+FTC/TDF, good tolerability profile, and high adherence. After discontinuation, EFV has an extended inductive effect on cytochrome P450 (CYP) 3A4 that, after switching, may reduce RPV exposures and adversely impact clinical outcomes. This study examines the clinical implications of reduced RPV exposures with concomitant FTC/TDF and declining EFV exposures when patients, intolerant to EFV, switch from EFV/FTC/TDF to RPV/FTC/TDF. This 48-week, phase 2b, open-label, multicenter study evaluated the efficacy and safety of switching from EFV/FTC/TDF (>/= 3 months duration) to RPV/FTC/TDF. Virologic suppression (HIV-1 RNA <50 copies/mL), safety, and EFV and RPV pharmacokinetics were assessed. At weeks 12 and 24, all 49 dosed subjects remained suppressed on RPV/FTC/TDF. At week 48, 46 (93.9%) subjects remained suppressed and virologic failure occurred in 2/49 (4.1%) subjects with no emergence of resistance. EFV concentrations were above the 90th percentile for inhibitory concentration (IC90) for several weeks after EFV discontinuation, and RPV exposures were in the range observed in phase 3 studies by approximately 2 weeks post switch. No subjects discontinued the study due to an adverse event. Switching from EFV/FTC/TDF to RPV/FTC/ TDF was a safe, efficacious option for virologically suppressed HIV-infected patients with EFV intolerance wishing to remain on an STR. Drug Warnings Adverse effects of moderate or severe intensity and reported in 2% or more of patients receiving rilpivirine include depressive disorders, insomnia, headache, and rash. Increased serum AST and/or ALT concentrations (more than 2.5 times the upper limit of normal (ULN) were reported in 3-4% of patients receiving rilpivirine. Rilpivirine should be used with caution and with increased monitoring for adverse effects in patients with severe renal impairment or end-stage renal disease since concentrations of the drug may be increased due to alterations in absorption, distribution, or metabolism. Rilpivirine and the fixed combination containing rilpivirine, emtricitabine, and tenofovir (Complera) have not been studied in patients with severe hepatic impairment (Child-Pugh class C). Experience in those 65 years of age and older is insufficient to determine whether they respond differently than younger adults. Dosage should be selected with caution because of age-related decreases in hepatic and/or renal function and potential for concomitant disease and drug therapy. For more Drug Warnings (Complete) data for Rilpivirine (13 total), please visit the HSDB record page. Pharmacodynamics Rilpivirine is a non-nucleoside reverse transcriptase inhibitor that inhibits the replication of HIV-1. It has a long duration of action as the oral tablet is given daily and the intramuscular suspension is given monthly. Patients should be counselled regarding the risk of hypersensitivity reactions, hepatotoxicity, depressive disorders, and the redistribution or accumulation of body fat. |
| 分子式 |
C22H18N6
|
|
|---|---|---|
| 分子量 |
366.42
|
|
| 精确质量 |
366.159
|
|
| CAS号 |
500287-72-9
|
|
| 相关CAS号 |
Rilpivirine hydrochloride;700361-47-3;Rilpivirine-d6;1312424-26-2
|
|
| PubChem CID |
6451164
|
|
| 外观&性状 |
Light yellow to yellow solid powder
|
|
| 密度 |
1.3±0.1 g/cm3
|
|
| 沸点 |
634.1±65.0 °C at 760 mmHg
|
|
| 熔点 |
245ºC
|
|
| 闪点 |
337.3±34.3 °C
|
|
| 蒸汽压 |
0.0±1.9 mmHg at 25°C
|
|
| 折射率 |
1.665
|
|
| LogP |
3.63
|
|
| tPSA |
97.42
|
|
| 氢键供体(HBD)数目 |
2
|
|
| 氢键受体(HBA)数目 |
6
|
|
| 可旋转键数目(RBC) |
5
|
|
| 重原子数目 |
28
|
|
| 分子复杂度/Complexity |
607
|
|
| 定义原子立体中心数目 |
0
|
|
| SMILES |
CC1=CC(=CC(=C1NC2=NC(=NC=C2)NC3=CC=C(C=C3)C#N)C)/C=C/C#N
|
|
| InChi Key |
YIBOMRUWOWDFLG-ONEGZZNKSA-N
|
|
| InChi Code |
InChI=1S/C22H18N6/c1-15-12-18(4-3-10-23)13-16(2)21(15)27-20-9-11-25-22(28-20)26-19-7-5-17(14-24)6-8-19/h3-9,11-13H,1-2H3,(H2,25,26,27,28)/b4-3+
|
|
| 化学名 |
4-{[4-({4-[(E)-2-cyanovinyl]-2,6-dimethylphenyl}amino)pyrimidin-2-yl]amino}benzonitrile
|
|
| 别名 |
|
|
| HS Tariff Code |
2934.99.9001
|
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
|
|||
|---|---|---|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 3 mg/mL (8.19 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 30.0 mg/mL 澄清的 DMSO 储备液加入到400 μL PEG300中,混匀;再向上述溶液中加入50 μL Tween-80,混匀;然后加入450 μL 生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: 3 mg/mL (8.19 mM) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 悬浊液; 超声助溶。 例如,若需制备1 mL的工作液,可将 100 μL 30.0 mg/mL澄清DMSO储备液加入900 μL 20% SBE-β-CD生理盐水溶液中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 View More
配方 3 中的溶解度: ≥ 3 mg/mL (8.19 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.7291 mL | 13.6455 mL | 27.2911 mL | |
| 5 mM | 0.5458 mL | 2.7291 mL | 5.4582 mL | |
| 10 mM | 0.2729 mL | 1.3646 mL | 2.7291 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
 NNRT-IN-5
NNRT-IN-5
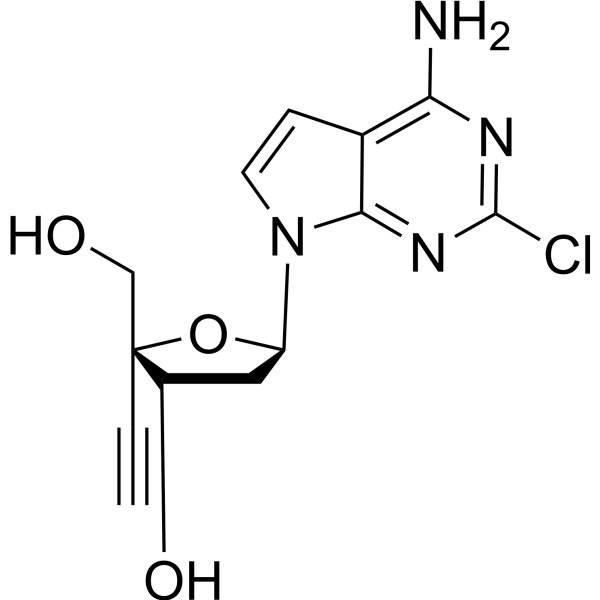 MK-8527
MK-8527
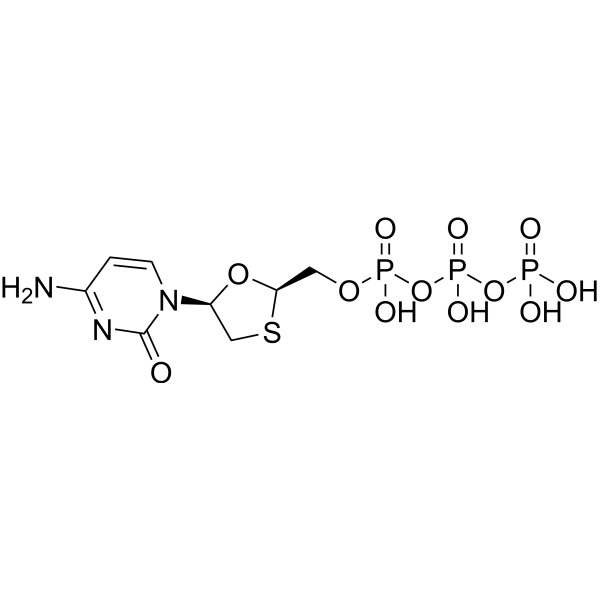 Lamivudine triphosphate
Lamivudine triphosphate
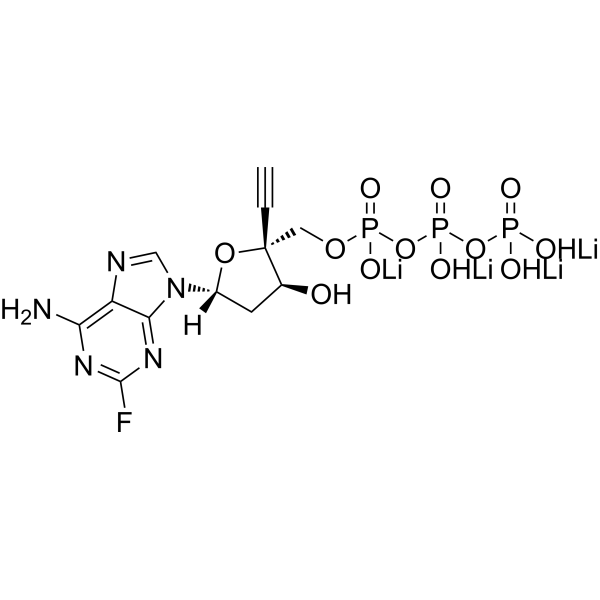 EFdA-TP tetralithium
EFdA-TP tetralithium
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1





























 COA
COA
