
| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 10 mM * 1 mL in DMSO |
|
||
| 1mg |
|
||
| 2mg |
|
||
| 5mg |
|
||
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| 500mg |
|
||
| Other Sizes |
|

| 靶点 |
Axl kinase (IC50 = 14 nM)
|
|---|---|
| 体外研究 (In Vitro) |
体外活性:R428 (也称为 BGB324)阻断 Axl 的催化和促癌活性。 R428 以低纳摩尔活性抑制 Axl,并阻断 Axl 依赖性事件,包括 Akt 磷酸化、乳腺癌细胞侵袭和促炎细胞因子产生。在最近的一项研究中,Axl 抑制剂 R428 显示治疗 24 小时后对原代 CLL B 细胞的平均 IC50 剂量约为 2.0μM,而正常 B、T 和自然杀伤 (NK) 细胞没有显示出显着的细胞数量在类似的实验条件下,该剂量的 R428 (2.5 μM) 导致死亡。激酶测定:R428(也称为 BGB324)是 Axl 的有效选择性抑制剂,IC50 为 14 nM,对 Axl 的选择性是 Abl 的 >100 倍。 R428 对 Axl 的选择性也高于 Mer 和 Tyro3(选择性高出 50 至 100 倍)以及 InsR、EGFR、HER2 和 PDGFRβ(选择性高出 100 倍)。在放射测定体外激酶测定中,在每种激酶的 KmATP 处对 133 种激酶进行了五点 R428 剂量滴定。 Axl、Mer 和 Tyro3 (Carna Biosciences) 测定也使用荧光偏振方案进行。 HER2 活性通过 Z-LYTE 测定法测定。细胞测定:将HeLa细胞接种于96孔板的饥饿培养基中。 24小时后,将细胞与稀释的R428预孵育1小时,然后用预聚集的抗Axl抗体刺激。将细胞固定、封闭并用抗磷酸 Akt (Ser473) 染色,然后用山羊抗兔辣根过氧化物酶染色,然后使用 SuperSignal ELISA Pico 化学发光底物进行显色。
|
| 体内研究 (In Vivo) |
药理学研究显示口服给药后有利的暴露,使得 R428 治疗的肿瘤细胞因子粒细胞巨噬细胞集落刺激因子和上皮间质转化转录调节因子 Snail 的表达出现剂量依赖性减少。为了支持一项早期研究,R428 抑制角膜微袋和肿瘤模型中的血管生成。 R428 给药可降低 MDA-MB-231 心内和 4T1 原位乳腺癌转移小鼠模型的转移负担并延长生存期(中位生存期 >80 天与 52 天相比;P < 0.05)。
|
| 酶活实验 |
体外激酶测定[1]
采用5点R428 剂量滴定法对133种激酶的KmATP进行了放射体外激酶测定。Axl、Mer和Tyro3的测定也采用荧光偏振法。用Z′-LYTE法测定HER2活性。 基于Axl细胞的检测方法[1] 将HeLa细胞接种于96孔板的饥饿培养基中。24小时后,细胞用稀释的R428 预孵育1小时,然后用预聚集抗axl抗体刺激。在SuperSignal ELISA Pico化学发光底物进行开发之前,将细胞固定、阻断并使用抗磷酸化akt (Ser473)和山羊抗兔辣根过氧化物酶进行染色。另见补充材料和方法。 R428 (也称为 BGB324)是一种强大的选择性 Axl 抑制剂,对 Axl 的选择性比 Abl 高 100 倍以上,IC50 为 14 nM。此外,R428 对 Axl 的选择性高于 Mer 和 Tyro3(选择性高出 50-100 倍)以及 InsR、EGFR、HER2 和 PDGFRβ(选择性高出 100 倍)。在体外放射激酶测定中,对 133 种激酶在每种激酶的 KmATP 上进行了五点 R428 剂量滴定。荧光偏振方案也用于 Axl、Mer 和 Tyro3测定。通过 Z-LYTE 测定,确定了 HER2 活性。 |
| 细胞实验 |
入侵检测[2]
将MDA-MB-231或4T1细胞(1 × 105)在孔径为8 μm的24孔Transwell板中,在37°C下通过Matrigel向20% FCS迁移16至24小时。通过拭子去除未受侵袭的细胞和Matrigel。浸润细胞用4%甲醛固定,1%结晶紫染色,定量用于Axl细胞检测。细胞用R428预孵育3小时。R428 分别加入上下两个Transwell室。 MDA-MB-231-luc-D3H2LN心内模型[2] 7 ~ 8周龄雌性NCr nu/nu小鼠(Taconic)心内注射生物发光MDA-MB-231-luc-D3H2LN细胞悬液。在细胞植入前2小时开始口服R428 (125 mg/kg)或对照剂,每天两次,持续到第21天(n = 20)。在第22天通过体内生物发光成像量化转移负荷,并使用Wilcoxon秩和检验进行分析。 在无血清培养基中培养 24 小时后,取出细胞并添加到 24 孔室的上室(每孔 1.5 x 10^5 个细胞),该上室可以是未包被(用于迁移)或包被有基质胶(用于入侵)。下室充满10%胎牛血清RPMI培养基。在将细胞装入上室之前,用载体(DMSO,0.25%)或 bemcentinib (R428 ) (2 μM) 处理两个小时。药物或媒介物存在于上室和下室中。使用 Infinite M1000 酶标仪上的 480/520 nm 滤光片组,分别在 20 小时或 42 小时后测量迁移或入侵细胞的荧光信号。 |
| 动物实验 |
Female NCr nu/nu mice aged seven to eight weeks are given intracardial injections of bioluminescent MDA-MB-231-luc-D3H2LN cell suspension. Two hours prior to cell implantation, oral dosage of Bemcentinib (R428) (125 mg/kg, p.o.) or vehicle is administered twice daily until day 21 (n=20). Day 22 in vivo bioluminescence imaging is used to measure the metastatic burden, and the Wilcoxon rank sum test is used for analysis.
Orthotopic Model[2] Female BALB/c mice were inoculated in the mammary fat pad with 0.5 × 106 4T1 cells. Forty-eight hours after inoculation, mice were randomized into treatment groups (n = 10). Oral dosing with R428 (7–75 mg/kg twice daily) or vehicle continued until days 19 to 21. Cisplatin (1.2 or 4 mg/kg) was administered i.v. once weekly. Body weight and tumor size were measured thrice per week. Lungs were exposed postmortem. Total number and size of surface lung macrometastases were measured (small, <2 mm; medium, ≥2 mm and <3 mm; large, ≥3 mm). Half of each primary tumor was snap frozen in liquid nitrogen. The other half, and the livers, were fixed in paraformaldehyde/lysine/periodate solution, paraffin embedded, and sectioned (5 μm thick). Two H&E-stained liver sections per animal were examined microscopically for micrometastases in three view fields. Synergism was determined using Clark's synergy calculation. |
| 参考文献 |
|
| 其他信息 |
Bemcentinib has been investigated for the treatment of Non-Small Cell Lung Cancer.
Bemcentinib is an orally available and selective inhibitor of the AXL receptor tyrosine kinase (UFO), with potential antineoplastic activity. Upon administration, bemcentinib targets and binds to the intracellular catalytic kinase domain of AXL and prevents its activity. This blocks AXL-mediated signal transduction pathways and inhibits the epithelial-mesenchymal transition (EMT), which, in turn, inhibits tumor cell proliferation and migration. In addition, bemcentinib enhances chemo-sensitivity. AXL, a member of the TAM (TYRO3, AXL and MER) family of receptor tyrosine kinases overexpressed by many tumor cell types, plays a key role in tumor cell proliferation, survival, invasion and metastasis; its expression is associated with drug resistance and poor prognosis. Axl, a member of the TAM (Tyro3, Axl, Mer) family of receptor tyrosine kinases, displays an increasingly important role in carcinogenesis. Analysis of 58 cutaneous melanoma lines indicated that Axl was expressed in 38% of them, with significant overrepresentation in NRAS- compared with BRAF-mutated tumors. Axl activation could be induced by autocrine production of its ligand, Gas6, in a significant fraction of Axl-positive tumors. Pearson's correlation analysis on expression data from five data sets of melanoma lines identified several transcripts correlating positively or negatively with Axl. By functionally grouping genes, those inversely correlated were involved in melanocyte development and pigmentation, whereas those positively correlated were involved in motility, invasion, and microenvironment interactions. Accordingly, Axl-positive melanomas did not express microphthalmia transcription factor (MITF) and melanocyte differentiation antigens (MDAs) such as MART-1 and gp100 and possessed a greater in vitro invasive potential compared with Axl-negative ones. Motility, invasivity, and ability to heal a wound or to migrate across an endothelial barrier were inhibited in vitro by Axl knockdown. Pharmacological inhibition of Axl using the selective inhibitor R428 had comparable effects in reducing migration and invasion. These results suggest that targeted inhibition of Axl signaling in the subset of melanomas lacking MITF and MDAs may represent a novel therapeutic strategy.[1] Accumulating evidence suggests important roles for the receptor tyrosine kinase Axl in cancer progression, invasion, metastasis, drug resistance, and patient mortality, highlighting Axl as an attractive target for therapeutic development. We have generated and characterized a potent and selective small-molecule inhibitor, R428, that blocks the catalytic and procancerous activities of Axl. R428 inhibits Axl with low nanomolar activity and blocked Axl-dependent events, including Akt phosphorylation, breast cancer cell invasion, and proinflammatory cytokine production. Pharmacologic investigations revealed favorable exposure after oral administration such that R428-treated tumors displayed a dose-dependent reduction in expression of the cytokine granulocyte macrophage colony-stimulating factor and the epithelial-mesenchymal transition transcriptional regulator Snail. In support of an earlier study, R428 inhibited angiogenesis in corneal micropocket and tumor models. R428 administration reduced metastatic burden and extended survival in MDA-MB-231 intracardiac and 4T1 orthotopic (median survival, >80 days compared with 52 days; P < 0.05) mouse models of breast cancer metastasis. Additionally, R428 synergized with cisplatin to enhance suppression of liver micrometastasis. Our results show that Axl signaling regulates breast cancer metastasis at multiple levels in tumor cells and tumor stromal cells and that selective Axl blockade confers therapeutic value in prolonging survival of animals bearing metastatic tumors.[2] A low-molecular-weight receptor tyrosine kinase inhibitor, 1-(6,7-dihydro-5H-benzo(6,7)cyclohepta(1,2-c)pyridazin-3-yl)-N3-((7-pyrrolidin-1-yl)-6,7,8,9-tetrahydro-5H-benzo(7)annulene-2-yl)-1H-1,2,4-triazole-3,5-diamine (R428) with high affinity and selectivity for the growth arrest-specific protein 6 (GAS6) receptor Axl was used to study a potential role of GAS6 signaling in adiposity. In vitro, R428 caused a concentration-dependent inhibition of preadipocyte differentiation into mature adipocytes, as evidenced by reduced lipid uptake. Inhibition of Axl-mediated signaling was confirmed by reduced levels of phospho-Akt activity. In vivo, oral administration of R428 for 5 weeks to mice kept on a high-fat diet resulted in significantly reduced weight gain and subcutaneous and gonadal fat mass. This was associated with marked adipocyte hypotrophy, enhanced macrophage infiltration, and apoptosis. Thus, affecting GAS6 signaling through receptor antagonism using a low-molecular-weight Axl antagonist impairs adipocyte differentiation and reduces adipose tissue development in a murine model of nutritionally induced obesity.[3] |
| 分子式 |
C30H34N8
|
|
|---|---|---|
| 分子量 |
506.64
|
|
| 精确质量 |
506.29
|
|
| 元素分析 |
C, 71.12; H, 6.76; N, 22.12
|
|
| CAS号 |
1037624-75-1
|
|
| 相关CAS号 |
1037624-75-1; 1037624-91-1 (racemic);
|
|
| PubChem CID |
46215462
|
|
| 外观&性状 |
Off-white to yellow solid powder
|
|
| 密度 |
1.4±0.1 g/cm3
|
|
| 沸点 |
799.6±70.0 °C at 760 mmHg
|
|
| 闪点 |
437.4±35.7 °C
|
|
| 蒸汽压 |
0.0±2.8 mmHg at 25°C
|
|
| 折射率 |
1.768
|
|
| LogP |
4.55
|
|
| tPSA |
98.51
|
|
| 氢键供体(HBD)数目 |
2
|
|
| 氢键受体(HBA)数目 |
7
|
|
| 可旋转键数目(RBC) |
4
|
|
| 重原子数目 |
38
|
|
| 分子复杂度/Complexity |
775
|
|
| 定义原子立体中心数目 |
1
|
|
| SMILES |
N1(C([H])([H])C([H])([H])C([H])([H])C1([H])[H])[C@@]1([H])C([H])([H])C([H])([H])C2C([H])=C([H])C(=C([H])C=2C([H])([H])C1([H])[H])N([H])C1N=C(N([H])[H])N(C2C([H])=C3C(C4=C([H])C([H])=C([H])C([H])=C4C([H])([H])C([H])([H])C3([H])[H])=NN=2)N=1
|
|
| InChi Key |
KXMZDGSRSGHMMK-VWLOTQADSA-N
|
|
| InChi Code |
InChI=1S/C30H34N8/c31-29-33-30(32-24-13-10-20-11-14-25(15-12-22(20)18-24)37-16-3-4-17-37)36-38(29)27-19-23-8-5-7-21-6-1-2-9-26(21)28(23)35-34-27/h1-2,6,9-10,13,18-19,25H,3-5,7-8,11-12,14-17H2,(H3,31,32,33,36)/t25-/m0/s1
|
|
| 化学名 |
1-(3,4-diazatricyclo[9.4.0.02,7]pentadeca-1(15),2,4,6,11,13-hexaen-5-yl)-3-N-[(7S)-7-pyrrolidin-1-yl-6,7,8,9-tetrahydro-5H-benzo[7]annulen-3-yl]-1,2,4-triazole-3,5-diamine
|
|
| 别名 |
|
|
| HS Tariff Code |
2934.99.9001
|
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
|
|||
|---|---|---|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: 2.08 mg/mL (4.11 mM) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 悬浮液;超声助溶。
例如,若需制备1 mL的工作液,可将100 μL 20.8 mg/mL澄清DMSO储备液加入900 μL 20% SBE-β-CD生理盐水溶液中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 配方 2 中的溶解度: 5% DMSO+corn oil: 1 mg/mL View More
配方 3 中的溶解度: 12.5 mg/mL (24.67 mM) in 0.5% CMC-Na/saline water (这些助溶剂从左到右依次添加,逐一添加), 悬浊液; 超声助溶。 配方 4 中的溶解度: 5 mg/mL (9.87 mM) in 0.5%HPMC 1%Tween80 (这些助溶剂从左到右依次添加,逐一添加), 悬浊液; 超声助溶。 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.9738 mL | 9.8689 mL | 19.7379 mL | |
| 5 mM | 0.3948 mL | 1.9738 mL | 3.9476 mL | |
| 10 mM | 0.1974 mL | 0.9869 mL | 1.9738 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
|
") |
") |
A, experimental protocol for MDA-MB-231-luc-D3H2LN metastasis prevention study.Cancer Res.2010 Feb 15;70(4):1544-54. |
A, experimental protocol for 4T1 orthotopic metastasis study.Cancer Res.2010 Feb 15;70(4):1544-54. |
R428 modulates expression of Snail and GM-CSF in 4T1 primary tumors.Cancer Res.2010 Feb 15;70(4):1544-54. |
 Denfivontinib hydrochloride
Denfivontinib hydrochloride
 (Z)-S49076 hydrochloride
(Z)-S49076 hydrochloride
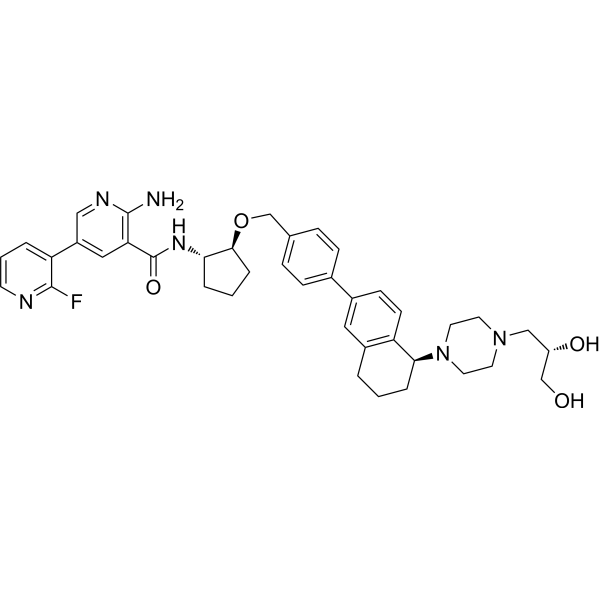 MerTK/Axl-IN-1
MerTK/Axl-IN-1
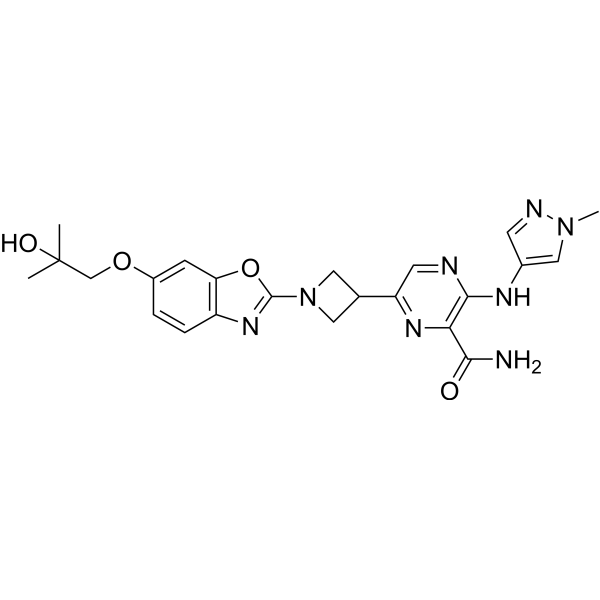 MerTK-IN-1
MerTK-IN-1
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1





























 COA
COA
")
")
")
")
