| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 10mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 500mg |
|
||
| 1g |
|
||
| Other Sizes |
|
| 靶点 |
c-Met (IC50 = 4 nM)
|
|---|---|
| 体外研究 (In Vitro) |
盐酸替泊替尼抑制 IRAK4、TrkA、Axl、IRAK1、Mer 和 TrkA,IC50 分别为 615、1017、1566、2037、2272 和 5716 nM[1]。盐酸替泊替尼抑制 A549 细胞中 HGF 诱导的 c-Met 磷酸化,平均 IC50 为 6 nM[1]。盐酸替泊替尼 (0.01 nM-30 μM) 可在体外抑制肿瘤细胞增殖和迁移[1]。
|
| 体内研究 (In Vivo) |
盐酸替泊替尼在异种移植模型中诱导肿瘤消退,并在体内抑制 c-Met 磷酸化 [1]。
|
| 酶活实验 |
EMD 1214063和EMD 1204831选择性地抑制c-Met受体酪氨酸激酶活性。与它们对242种人类激酶的作用相比,它们的抑制活性是有效的[分别为抑制50%浓度(IC50)、3 nmol/L和9 nmol/L]和高度选择性的。EMD 1214063和EMD 1204831均以剂量依赖的方式抑制c-Met磷酸化和下游信号传导,但其抑制活性的持续时间不同。[1]
c-Met体外激酶测定[1] 使用242种不同的激酶在体外评估EMD 1214063或EMD 1204831(1和10μmol/L)对激酶的抑制作用。用闪板法测定生物化学活性。将His6标记的重组人c-Met激酶结构域(Aa 974–end;20 ng)和生物素化的聚Ala-Glu-Lys-Tyr(6:2:5:1;500 ng)在室温下与测试化合物一起或不与测试化合物在100μL缓冲液中孵育90分钟,该缓冲液含有0.3μCi 33P-ATP、2.5μg聚乙二醇20.000和1%二甲基亚砜(DMSO),如前所述。用TopCount微孔板闪烁和发光计数器测量放射性。使用RS/1软件程序通过非线性回归分析计算抑制50%浓度值(IC50)。[1] 磷酸化c-Met捕获ELISA[1] 在Nunc Immuno MicroWell 96孔固体板中通过c-Met捕获ELISA评估总的c-Met磷酸化。A549人癌症细胞在治疗前2天接种,血清饥饿20小时,并在第3天用不同浓度的EMD 1214063或EMD 1204831或0.2%DMSO在37°C、5%CO2下治疗45分钟。在用100 ng/mL HGF刺激5分钟后,用补充有蛋白酶和磷酸酶抑制剂的70μL/孔冰冷裂解缓冲液[20 nmol/L HEPES,pH 7,4;10%(V/V)甘油;150 nmol/L NaCl;1%(V/V的)Triton-X-100;2 nmol/L EDTA]裂解细胞。在冲洗实验中,A549用EMD 1214063或EMD 1204831处理45分钟,洗涤,并在无血清培养基中孵育14小时,然后用HGF(100ng/mL)刺激。 在ELISA中,捕获抗体对c-Met细胞外结构域是特异性的,而抗磷脂-生物素标记的抗体用于检测。使用链霉亲和素-过氧化物酶缀合物和化学发光读数揭示酪氨酸磷酸化。 生化分析[1] 在EBC-1细胞中通过蛋白质印迹分析分析c-Met、Gab-1、Akt和Erk1/2的磷酸化。简言之,细胞以每孔3×106个细胞的密度接种,血清饥饿20小时,并在与EMD 1214063孵育后的第3天裂解。通过SDS-PAGE分离蛋白质并将其吸附在硝化纤维素膜上。用Tris缓冲盐水封闭膜,并在4°C的一级抗体溶液(抗pMet、抗pAkt、抗pERK1/2、抗Gab1)中孵育过夜。用装有Quantity One 1-D分析软件的VersaDoc MP 5000成像系统通过化学发光检测蛋白质。 |
| 细胞实验 |
Tepotinib (EMD-1214063) 是一种有效且具有选择性的 c-Met 抑制剂。它的 IC50 为 4 nM,对 c-Met 的选择性比 IRAK4、TrkA、Axl、IRAK1 和 Mer 的选择性高 200 倍以上。
伤口愈合试验和增殖试验[1] 伤口愈合试验如前所述进行。简言之,用无菌移液管尖端在单层NCI-H441癌症细胞上产生划痕。在存在或不存在100ng/mL HGF的情况下,在24小时内监测EMD 1210463和EMD 1204831对细胞间隙闭合的影响。所有增殖和集落形成测定在4个重复中进行,并包括4个DMSO载体对照。IC50值通过GraphPad Prism v5中的4PL拟合确定。 离体肿瘤样品的药效学标志物[1] 通过对冷冻离体肿瘤样品的蛋白质印迹分析来研究c-Met自磷酸化。根据制造商的说明,使用Precellys 24均质器或Precellys陶瓷裂解管(PEQLab Ltd)对肿瘤组织进行机械均质、裂解。裂解物的进一步制备和通过SDS-PAGE的蛋白质分离如已经对EBC-1细胞描述的那样进行。 在福尔马林固定的石蜡包埋切片上,通过免疫组织化学(IHC)分析组蛋白H3磷酸化和细胞周期停滞和凋亡的生物标志物(细胞周期蛋白D1、p27和裂解的活化的capase-3)。根据制造商的说明,使用Discovery染色仪器和OmniMap试剂盒进行IHC。切片用苏木精复染。 |
| 动物实验 |
Animal/Disease Models:CD-1 or BALB/C nude mice bearing human cancer cell lines KP-4, or EBC-1[1]
Doses: 6 and 15 mg/kg for mice bearing NSCLC EBC-1; 25, 50 and 200 mg/kg for mice bearing pancreatic carcinoma cell line KP-4. Route of Administration: Injected daily; for 14-18 days Experimental Results: Daily administration of 5 or 15 mg/kg to EBC-1 tumor-bearing mice resulted in effective inhibition or complete tumor regression, respectively. Induced dose-dependent tumor growth inhibition in mice bearing human pancreatic carcinoma KP-4 tumors. The antitumor efficacy of EMD 1214063 or EMD 1204831 was investigated in mouse xenograft models. CD-1 or BALB/C nude mice were injected subcutaneously with human cancer cell lines KP-4, U87MG: 10 × 106 cells in 100 μL, Hs746T, EBC-1: 5 × 106 cells in 100 μL. As soon as the tumor reached the linear growth phase (70–150 mm3), tumor-bearing mice (10 mice/group) were injected daily with the indicated doses of EMD 1214063 or EMD 1204831, or vehicle. Body weight and tumor size [length (L) and width (W)] were measured twice weekly. The tumor volume was calculated using the formula L × W2/2. Statistical significance was determined by one-way ANOVA. P ≤ 0.05 were considered significant. Pharmacokinetic and pharmacodynamic studies[1] Plasma and tumor drug concentrations were measured using high-performance liquid chromatography (HPLC) and mass spectrometry (MS). In brief, protein precipitation was carried out in methanol for plasma samples, and in ethanol/water 80:20 (v/v) using a Precellys 24 homogenizer for homogenized tumor samples. The HPLC/tandem mass spectrometry (MS-MS) system consisted of an Agilent 1100 Series HPLC system with a CTC HTC PAL Autosampler coupled to an Applied Biosystems API4000 mass spectrometer. HPLC separation was achieved on a reversed-phase column (Chromolith SpeedROD RP-18e, 50–3 mm) using gradient elution (eluent A: formic acid 0.1%; eluent B: acetonitrile). Selectivity was achieved using multiple reaction monitoring (MRM) for the MS/MS detection of the compounds. For the in vivo pharmacodynamic studies, all animal studies were conducted according to standard procedures approved by local animal welfare authorities. Mice were injected subcutaneously with 5 × 106 Hs746T cells (100 μL). Once the tumor volume had reached 600 to 1,000 mm3, mice were randomized into different experimental groups, receiving a single oral dose of 3, 10, 30, and 100 mg/kg of EMD 1214063, EMD 1204831, or vehicle. Tumor and plasma samples were collected at 3, 6, 12, 24, 48, 72, and 96 hours after treatment. Each experimental group comprised 4 mice per dose and time point. Samples of the tumor tissue were snap-frozen for pharmacokinetic and biomarker analyses, or formalin-fixed for immunohistochemical analysis. |
| 药代性质 (ADME/PK) |
Absorption
The absolute bioavailability of tepotinib following oral administration is approximately 72%. At the recommended dosage of 450mg once daily, the median Tmax is 8 hours and the mean steady-state Cmax and AUC0-24h were 1,291 ng/mL and 27,438 ng·h/mL, respectively. Co-administration with a high-fat, high-calorie meal increases the AUC and Cmax of tepotinib by approximately 1.6-fold and 2-fold, respectively. Route of Elimination Following oral administration, approximately 85% of the given dose is excreted in the feces with the remainder excreted in the urine. Unchanged parent drug accounts for roughly half of the dose excreted in the feces, with the remainder comprising the demethylated M478 metabolite, a glucuronide metabolite, the racemic M506 metabolite, and some minor oxidative metabolites. Unchanged parent drug also accounts for roughly half of the dose excreted in the urine, with the remainder comprising a glucuronide metabolite and a pair of N-oxide diastereomer metabolites. Volume of Distribution The mean apparent volume of distribution is 1,038L. Clearance The apparent clearance of tepotinib is 23.8 L/h. Metabolism / Metabolites Tepotinib is metabolized primarily by CYP3A4 and CYP2C8, with some apparent contribution by unspecified UGT enzymes. The metabolite M506 is the major circulating metabolite, comprising approximately 40.4% of observed drug material in plasma, while the M668 glucuronide metabolite has been observed in plasma at much lower quantities (~4% of an orally administered dose). A total of 10 phase I and phase II metabolites have been detected following tepotinib administration, most of which are excreted in the feces. Biological Half-Life Following oral administration, the half-life of tepotinib is approximately 32 hours. |
| 毒性/毒理 (Toxicokinetics/TK) |
Hepatotoxicity
In the prelicensure clinical trials of tepotinib in patients with solid tumors harboring MET mutations, liver test abnormalities were frequent although usually self-limited and mild. Some degree of ALT elevations arose in 44% of tepotinib treated patients and were above 5 times the upper limit of normal (ULN) in 4%. In these trials that enrolled 255 patients, dose interruptions due to ALT or AST elevations occurred in 3%, but permanent discontinuations in less than 1%. The liver test abnormalities had a median onset of 30 days after initiation of therapy. While serum aminotransferase elevations were occasionally quite high (5 to 20 times upper limit of normal), there were no accompanying elevations in serum bilirubin and no patient developed clinically apparent liver injury with jaundice. The product label for tepotinib recommends monitoring for routine liver tests before, at 2 week intervals during the first 3 months of therapy, and monthly thereafter as clinically indicated. Likelihood score: E* (unproven but suspected rare cause of clinically apparent liver injury). Effects During Pregnancy and Lactation ◉ Summary of Use during Lactation No information is available on the clinical use of tepotinib during breastfeeding. Because tepotinib is 98% bound to plasma proteins, the amount in milk is likely to be low. However, because of its potential toxicity in the breastfed infant and its half-life of 32 hours, the manufacturer recommends that breastfeeding be discontinued during tepotinib therapy and for 1 week after the last dose. ◉ Effects in Breastfed Infants Relevant published information was not found as of the revision date. ◉ Effects on Lactation and Breastmilk Relevant published information was not found as of the revision date. Protein Binding Tepotinib is approximately 98% protein-bound in plasma, primarily to serum albumin and alpha-1-acid glycoprotein. Plasma protein binding is independent of drug concentration at clinically relevant exposures. |
| 参考文献 |
|
| 其他信息 |
See also: Tepotinib Hydrochloride (annotation moved to).
|
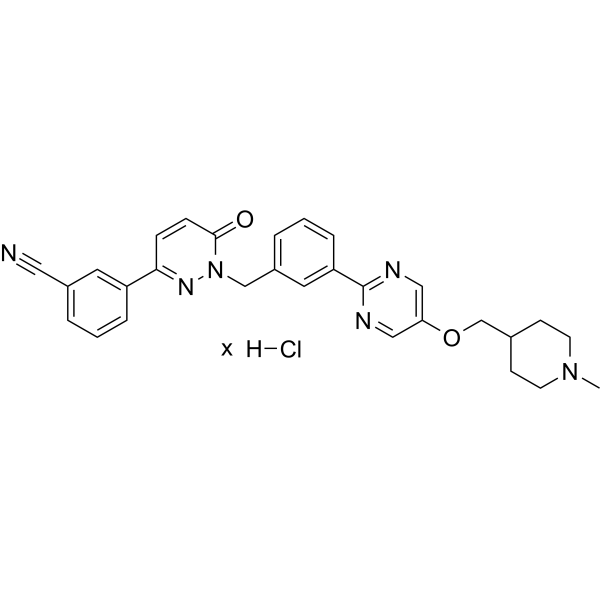
| 分子式 |
C29H28N6O2.XHCL
|
|---|---|
| 分子量 |
492.57 (free base)
|
| 精确质量 |
528.204
|
| CAS号 |
1103508-80-0
|
| 相关CAS号 |
1100598-32-0;1946826-82-9
|
| PubChem CID |
25171647
|
| 外观&性状 |
Typically exists as solid at room temperature
|
| tPSA |
94.7
|
| 氢键供体(HBD)数目 |
1
|
| 氢键受体(HBA)数目 |
7
|
| 可旋转键数目(RBC) |
7
|
| 重原子数目 |
38
|
| 分子复杂度/Complexity |
880
|
| 定义原子立体中心数目 |
0
|
| SMILES |
C(C1C=CC=C(C2=NC=C(OCC3CCN(C)CC3)C=N2)C=1)N1C(C=CC(C2C=CC=C(C#N)C=2)=N1)=O.Cl
|
| InChi Key |
YHHHGHDGBUUWIS-UHFFFAOYSA-N
|
| InChi Code |
InChI=1S/C29H28N6O2.ClH/c1-34-12-10-21(11-13-34)20-37-26-17-31-29(32-18-26)25-7-3-5-23(15-25)19-35-28(36)9-8-27(33-35)24-6-2-4-22(14-24)16-30;/h2-9,14-15,17-18,21H,10-13,19-20H2,1H3;1H
|
| 化学名 |
3-[1-[[3-[5-[(1-methylpiperidin-4-yl)methoxy]pyrimidin-2-yl]phenyl]methyl]-6-oxopyridazin-3-yl]benzonitrile;hydrochloride
|
| 别名 |
EMD-1214063 hydrochloride; 8B73AZL5XP; 1103508-80-0; Tepotinib hydrochloride anhydrous; UNII-8B73AZL5XP; Benzonitrile, 3-(1,6-dihydro-1-((3-(5-((1-methyl-4-piperidinyl)methoxy)-2-pyrimidinyl)phenyl)methyl)-6-oxo-3-pyridazinyl)-, hydrochloride (1:1); SCHEMBL1295616; YHHHGHDGBUUWIS-UHFFFAOYSA-N; 3-(1-{3-[5-(1-methyl-piperidin-4-ylmethoxy)-pyrimidin-2-yl]-benzyl}-6-oxo-1,6-dihydro-pyridazin-3-yl)-benzonitrile hydrochloride;
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples
|
|---|---|
| 溶解度 (体内实验) |
注意: 如下所列的是一些常用的体内动物实验溶解配方,主要用于溶解难溶或不溶于水的产品(水溶度<1 mg/mL)。 建议您先取少量样品进行尝试,如该配方可行,再根据实验需求增加样品量。
注射用配方
注射用配方1: DMSO : Tween 80: Saline = 10 : 5 : 85 (如: 100 μL DMSO → 50 μL Tween 80 → 850 μL Saline)(IP/IV/IM/SC等) *生理盐水/Saline的制备:将0.9g氯化钠/NaCl溶解在100 mL ddH ₂ O中,得到澄清溶液。 注射用配方 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (如: 100 μL DMSO → 400 μL PEG300 → 50 μL Tween 80 → 450 μL Saline) 注射用配方 3: DMSO : Corn oil = 10 : 90 (如: 100 μL DMSO → 900 μL Corn oil) 示例: 以注射用配方 3 (DMSO : Corn oil = 10 : 90) 为例说明, 如果要配制 1 mL 2.5 mg/mL的工作液, 您可以取 100 μL 25 mg/mL 澄清的 DMSO 储备液,加到 900 μL Corn oil/玉米油中, 混合均匀。 View More
注射用配方 4: DMSO : 20% SBE-β-CD in Saline = 10 : 90 [如:100 μL DMSO → 900 μL (20% SBE-β-CD in Saline)] 口服配方
口服配方 1: 悬浮于0.5% CMC Na (羧甲基纤维素钠) 口服配方 2: 悬浮于0.5% Carboxymethyl cellulose (羧甲基纤维素) 示例: 以口服配方 1 (悬浮于 0.5% CMC Na)为例说明, 如果要配制 100 mL 2.5 mg/mL 的工作液, 您可以先取0.5g CMC Na并将其溶解于100mL ddH2O中,得到0.5%CMC-Na澄清溶液;然后将250 mg待测化合物加到100 mL前述 0.5%CMC Na溶液中,得到悬浮液。 View More
口服配方 3: 溶解于 PEG400 (聚乙二醇400) 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
| NCT Number | Recruitment | interventions | Conditions | Sponsor/Collaborators | Start Date | Phases |
| NCT03940703 | Active Recruiting |
Drug: Tepotinib Drug: Osimertinib |
Non-small Cell Lung Cancer | EMD Serono Research & Development Institute, Inc. |
September 19, 2019 | Phase 2 |
| NCT02864992 | Active Recruiting |
Drug: Tepotinib | Lung Adenocarcinoma Stage IIIB/IV Amplification |
EMD Serono Research & Development Institute, Inc. |
September 13, 2016 | Phase 2 |
| NCT05120960 | Recruiting | Drug: tepotinib plus osimertinib Drug: tepotinib |
Brain Tumor | M.D. Anderson Cancer Center | February 27, 2023 | Phase 1 |
| NCT04647838 | Recruiting | Drug: Tepotinib | Solid Tumor MET Amplification |
Chungbuk National University Hospital |
January 16, 2020 | Phase 2 |
| NCT05782361 | Recruiting | Drug: Tepotinib Drug: Pembrolizumab |
Non Small Cell Lung Cancer Advanced Cancer |
Institute of Cancer Research, United Kingdom |
May 3, 2023 | Phase 1 |
 Tepotinib hydrochloride monohydrate
Tepotinib hydrochloride monohydrate
 BKN-1
BKN-1
 NEO214
NEO214
 MJO445
MJO445
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1






























