| 规格 | 价格 | |
|---|---|---|
| 500mg | ||
| 1g | ||
| Other Sizes |

| 靶点 |
NMDA Receptor
|
|---|---|
| 体外研究 (In Vitro) |
DL-AP5 (100 μM) 可部分阻止谷氨酸诱导的 Arc/Arg3.1 蛋白水平升高[5]。DL-AP5 可降低 NMDA 诱导的 Arc/Arg3.1 上调[5]。
|
| 体内研究 (In Vivo) |
DL-AP5 (0-10 μg/大鼠,CA1 内注射) 显著降低 NMDA 的作用[3]。DL-AP5 (0-10 nmol,脑室内注射) 导致食物消耗呈剂量依赖性增加[4]。DL-AP5 (5 nmol,脑室内注射) 可减轻脑室内注射生长素释放肽引起的食物消耗减少[4]。
虽然许多证据表明P物质(SP),一种内源性神经激肽(NK),是哺乳动物脊髓急性疼痛的主要感觉递质,但其在持续(强直性)疼痛中的作用尚不清楚。虽然谷氨酸在背根神经节神经元中与SP共定位,但其在伤害性加工中的作用尚不确定。虽然在一些急性试验中发现NKs和兴奋性氨基酸(EAAs)的拮抗剂具有抗痛觉作用,但尚未对强直性疼痛进行过试验。我们推测:(1)NKs和EAAs参与补益性化学伤害感受的信号传导;(2) NK和EAA系统之间的相互作用是决定连续有害刺激感知强度的重要因素。因此,我们评估了两种NK拮抗剂([D-Pro2, d - trp7,9] SP(DPDT-SP, 0.26-6.6 nmol,非特异性)和[D-Pro4, d - trp7,9,10,Phe11]-SP(4-11) (DPDTP-octa, 1.6-12.3 nmol,有些NK-1选择性),以及dl -2-氨基-5-磷酸戊酸酯(DL-AP5, NMDA拮抗剂,0.05-1 nmol)和聚氨酯(一种kainic酸(KA)拮抗剂,2.5 mol)在小鼠福尔马林模型中的抗伤性活性。鞘内给药(i.t), DL-AP5和两种NK拮抗剂都具有显著的抗伤性,而聚氨酯(2.5摩尔)和纳洛酮(2.7摩尔)则无活性。平均镇痛百分比,nmol /小鼠i.t (95% CLs)的A50值为:DPDT-SP, 1.1 (0.79-1.6);DPDTP-octa, 3.9 (2.4-6.1);Dl-ap5, 0.29(0.16-0.71)。与1.3 nmol的DPDT-SP相关的抗疼痛作用未被2.7 nmol的纳洛酮逆转。0.1 nmol DL-AP5与1.3 nmol DPDT-SP或3.3 nmol dpdt -octa共给药均未产生加性抗痛作用[1]。 本试验旨在研究脑室内注射DL-AP5 (n-甲基-d -天冬氨酸(NMDA)受体拮抗剂)和谷氨酸对饥饿素诱导的3 h缺食(FD3)肉仔鸡摄食行为的影响。首先在鸡右侧脑室手术植入引导管。实验1,分别在脑室内注射0、2.5、5、10 nmol DL-AP5。实验2,在注射胃饥饿素0.6 nmol之前,先给鸡注射5 nmol DL-AP5。实验3,在谷氨酸300 nmol后,再饲喂0.6 nmol胃饥饿素,注射后3 h测定累积采食量。本研究结果表明,脑室内注射DL-AP5可增加FD3肉鸡的摄食量(P≤0.05),且呈剂量依赖性。谷氨酸预处理能增强脑室注射胃饥饿素引起的摄食量减少,DL-AP5能减弱这种作用(P≤0.05)。这些结果表明,胃饥饿素和谷氨酸能系统(通过NMDA受体)对肉仔鸡的摄食量有相互作用。[4] |
| 细胞实验 |
1. 对兔视网膜神经节细胞进行细胞内和细胞外记录。采用灌注给药方法研究n -甲基- dl -天冬氨酸(NMDLA)和n -甲基- d -天冬氨酸(NMDA)拮抗剂的作用。2. NMDLA刺激所有神经节细胞类型并引起特征性的突发放电模式,这在视网膜中不是典型的生理反应。当突触传递被钴阻断时,NMDLA仍然兴奋神经节细胞,表明直接作用。3. 比较dl -2-氨基-5-磷酸戊酸酯(DL-AP-5)和dl -2-氨基-7-磷酸庚酸酯(DL-AP-7)发现DL-AP-7是一种特异性更高的NMDA拮抗剂。DL-AP-5部分阻断视网膜电图(ERG)的b波,这是l -2-氨基-4-磷酸丁酸酯(L-APB)的典型作用,特异性阻断视网膜通道。4. DL-AP-7可逆转阻断NMDLA对所有神经节细胞类型的作用,但kainate (KA)和carbachol的作用不变。AP-7具有立体特异性和药理学特异性,在兔视网膜中具有竞争性NMDA拮抗剂的典型作用。5. DL-AP-7不阻断由中心或周围刺激驱动的ON或OFF神经节细胞的光反应。DL-AP-7对定向选择性没有影响。然而,大多数神经节细胞在光刺激下产生的动作电位数量减少,通常为20-30%。6. 与之前的报道相反,我们发现没有证据表明DL-AP-7特异性地抑制持续的ON神经节细胞。DL-AP-5对持续ON反应的抑制,以前被认为是NMDA的拮抗作用,可能是因为L-AP-5的APB活性较弱。7. 我们得出结论,NMDA受体不介导主要的光驱动输入到兔视网膜神经节细胞。通过排除,从双极细胞到神经节细胞的传播似乎主要由KA或准质(QQ)受体携带。然而,由于NMDA拮抗剂减少了光刺激产生的动作电位的数量,NMDA受体很可能携带部分信号传递到神经节细胞。NMDA受体在三阶神经元上的存在与谷氨酸从突触前神经元(如双极细胞)释放一致。[2]
|
| 动物实验 |
Animal/Disease Models: Male Wistar rats (180-230 g)[3]
Doses: 1, 3.2 and 10 μg/rat Route of Administration: Injected into the intra-dorsal hippocampal (intra-CA1) immediately after shock administration, once Experimental Results: Significantly decreased the effect of NMDA (10-2 μg/rat, intra-CA1) with significant interaction. Animal/Disease Models: Broilers cockerels (3-h fooddeprived (FD3), n=8 for each group)[4] Doses: 0, 2.5, 5, and 10 nmol; in a volume of 10 μL Route of Administration: Intracerebroventricular injection Experimental Results: Caused a dose-dependent increase in food consumption which was significant for 5 and 10 nmol doses. Animal/Disease Models: Broilers cockerels (3-h fooddeprived (FD3), n=8 for each group)[4] Doses: 5 nmol Route of Administration: Intracerebroventricular injection, followed by ghrelin (0.6 nmol) Experimental Results: Attenuated the decreased food consumption induced by the intracerebroventricular injection of ghrelin. |
| 参考文献 |
|
| 其他信息 |
In the present study, the effects of post-training intra-dorsal hippocampal (intra-CA1) injection of an N-methyl-D-aspartate (NMDA) receptor agonist and competitive or noncompetitive antagonists, on memory retention of passive avoidance learning was measured in the presence and absence of physostigmine in rats. Intra-CA1 administration of lower doses of the NMDA receptor agonist NMDA (10(-5) and 10(-4) microg/rat) did not affect memory retention, although the higher doses of the drug (10(-3), 10(-2) and 10(-1) microg/rat) increased memory retention. The greatest response was obtained with 10(-1) microg/rat of the drug. The different doses of the competitive NMDA receptor antagonist DL-AP5 (1, 3.2 and 10 microg/rat) and noncompetitive NMDA receptor antagonist MK-801 (0.5, 1 and 2 microg/rat) decreased memory retention in rats dose dependently. Both competitive and noncompetitive NMDA receptor antagonists reduced the effect of NMDA (10(-2) microg/rat). In another series of experiments, intra-CA1 injection of physostigmine (2, 3 and 4 microg/rat) improved memory retention. Post-training co-administration of lower doses of NMDA (10(-5) and 10(-4) microg/rat) and physostigmine (1 microg/rat), doses which were ineffective when given alone, significantly improved the retention latency. The competitive and noncompetitive NMDA receptor antagonists, DL-AP5 and MK-801, decreased the effect of physostigmine (2 microg/rat). Atropine decreased memory retention by itself and potentiated the response to DL-AP5 and MK-801. In conclusion, it seems that both NMDA and cholinergic systems not only play a part in the modulation of memory in the dorsal hippocampus of rats but also have demonstrated a complex interaction as well.[3]
Arc/Arg3.1 is a unique immediate early gene whose expression is highly dynamic and correlated with various forms of synaptic plasticity. Many previous reports highlight the complexity of mechanisms that regulate Arc/Arg3.1 expression in neurons. In the present study, the expression and regulation of Arc/Arg3.1 after glutamate treatment in primary cultured cortical neurons were investigated. We found that both Arc/Arg3.1 mRNA and Arc/Arg3.1 protein dynamically increased within 24h after glutamate treatment. The results of immunostaining showed that abundant amounts of Arc/Arg3.1 protein are presented in both soma and dendrites. The glutamate-induced increase in Arc/Arg3.1 protein levels was partially prevented by the NMDAR inhibitor DL-AP5, but not the AMPAR inhibitor NBQX. The results of calcium imaging showed that glutamate induced significant increases in intracellular calcium levels in a NMDAR-dependent manner. However, the intracellular calcium chelator BAPTA-AM had no effect on glutamate-induced upregulation of Arc/Arg3.1 protein, and alteration of cytosolic calcium ion homeostasis with A23187 and TG did not change Arc/Arg3.1 protein levels. In addition, the phosphorylation of ERK and CREB, two downstream factors of NMDAR signaling, markedly increased after glutamate exposure. Blocking ERK and CREB activation via selective inhibitors partially prevented the glutamate-induced elevation of Arc/Arg3.1 protein levels. Combined observations support a NMDAR-mediated and calcium-independent mechanism by which glutamate increases Arc/Arg3.1 expression in cortical neurons.[5] |
| 分子式 |
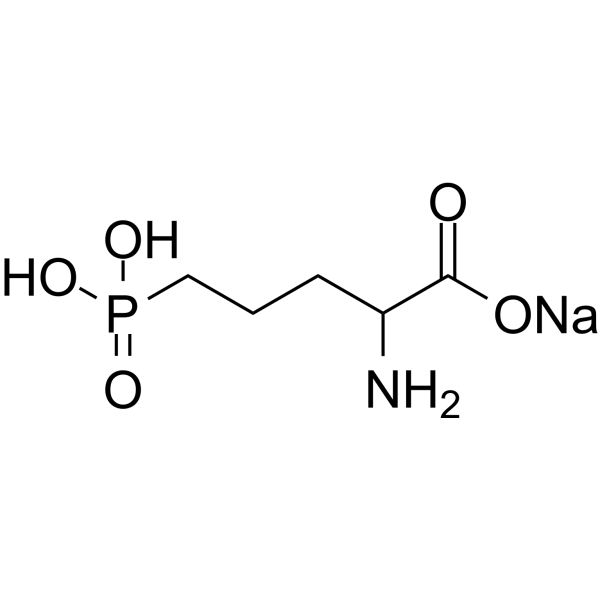
C5H11NNAO5P
|
|---|---|
| 分子量 |
219.11
|
| 精确质量 |
219.027
|
| CAS号 |
1303993-72-7
|
| PubChem CID |
52974251
|
| 外观&性状 |
White to off-white solid powder
|
| tPSA |
124
|
| 氢键供体(HBD)数目 |
3
|
| 氢键受体(HBA)数目 |
6
|
| 可旋转键数目(RBC) |
5
|
| 重原子数目 |
13
|
| 分子复杂度/Complexity |
211
|
| 定义原子立体中心数目 |
0
|
| InChi Key |
KWRCYAPNGUCHOE-UHFFFAOYSA-M
|
| InChi Code |
InChI=1S/C5H12NO5P.Na/c6-4(5(7)8)2-1-3-12(9,10)11;/h4H,1-3,6H2,(H,7,8)(H2,9,10,11);/q;+1/p-1
|
| 化学名 |
sodium;(4-amino-4-carboxybutyl)-hydroxyphosphinate
|
| 别名 |
DL-AP5 Sodium salt; 1303993-72-7; sodium;(4-amino-4-carboxybutyl)-hydroxyphosphinate; dl-2-amino-5-phosphonopentanoic acid sodium salt; D-LAP5.Na; C5H11NNaO5P;
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
Typically soluble in DMSO (e.g. 10 mM)
|
|---|---|
| 溶解度 (体内实验) |
注意: 如下所列的是一些常用的体内动物实验溶解配方,主要用于溶解难溶或不溶于水的产品(水溶度<1 mg/mL)。 建议您先取少量样品进行尝试,如该配方可行,再根据实验需求增加样品量。
注射用配方
注射用配方1: DMSO : Tween 80: Saline = 10 : 5 : 85 (如: 100 μL DMSO → 50 μL Tween 80 → 850 μL Saline)(IP/IV/IM/SC等) *生理盐水/Saline的制备:将0.9g氯化钠/NaCl溶解在100 mL ddH ₂ O中,得到澄清溶液。 注射用配方 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (如: 100 μL DMSO → 400 μL PEG300 → 50 μL Tween 80 → 450 μL Saline) 注射用配方 3: DMSO : Corn oil = 10 : 90 (如: 100 μL DMSO → 900 μL Corn oil) 示例: 以注射用配方 3 (DMSO : Corn oil = 10 : 90) 为例说明, 如果要配制 1 mL 2.5 mg/mL的工作液, 您可以取 100 μL 25 mg/mL 澄清的 DMSO 储备液,加到 900 μL Corn oil/玉米油中, 混合均匀。 View More
注射用配方 4: DMSO : 20% SBE-β-CD in Saline = 10 : 90 [如:100 μL DMSO → 900 μL (20% SBE-β-CD in Saline)] 口服配方
口服配方 1: 悬浮于0.5% CMC Na (羧甲基纤维素钠) 口服配方 2: 悬浮于0.5% Carboxymethyl cellulose (羧甲基纤维素) 示例: 以口服配方 1 (悬浮于 0.5% CMC Na)为例说明, 如果要配制 100 mL 2.5 mg/mL 的工作液, 您可以先取0.5g CMC Na并将其溶解于100mL ddH2O中,得到0.5%CMC-Na澄清溶液;然后将250 mg待测化合物加到100 mL前述 0.5%CMC Na溶液中,得到悬浮液。 View More
口服配方 3: 溶解于 PEG400 (聚乙二醇400) 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 4.5639 mL | 22.8196 mL | 45.6392 mL | |
| 5 mM | 0.9128 mL | 4.5639 mL | 9.1278 mL | |
| 10 mM | 0.4564 mL | 2.2820 mL | 4.5639 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
 17alpha-Hydroxywithanolide D
17alpha-Hydroxywithanolide D
 Lu AF90103
Lu AF90103
 Argiotoxin 636
Argiotoxin 636
 NMDA agonist 1
NMDA agonist 1
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1





























