| 规格 | 价格 | |
|---|---|---|
| 500mg | ||
| 1g | ||
| Other Sizes |

| 靶点 |
CDK7 (IC50 = 3.2 nM)
|
|---|---|
| 体外研究 (In Vitro) |
肿瘤癌基因包括选择一般转录机制来维持致癌状态的转录因子,但迄今为止,直接药理学抑制转录因子已被证明是困难的。然而,转录机制包含各种酶辅因子,可以靶向开发新的治疗候选物,包括细胞周期蛋白依赖性激酶(CDKs)。在这里,我们介绍了共价CDK7抑制剂THZ1的发现和表征,THZ1具有前所未有的靶向位于规范激酶结构域外的远程半胱氨酸残基的能力,为CDK7的选择性提供了一种意想不到的方法。癌症细胞系图谱表明,癌症细胞系的一个子集,包括人类T细胞急性淋巴细胞白血病(T-ALL),对THZ1具有异常的敏感性。Jurkat T-ALL细胞的全基因组分析表明,THZ1不成比例地影响RUNX1的转录,并表明对THZ1的敏感性可能是由于RUNX1超增强子赋予的脆弱性以及RUNX1在这些肿瘤细胞的核心转录调控回路中的关键作用。因此,CDK7激酶活性的药理学调节可能为鉴定和治疗依赖于转录维持致癌状态的肿瘤类型提供一种方法[1]。
|
| 酶活实验 |
抑制剂处理实验[1]
进行了扩展数据图5a中描述的时间过程实验,以确定CDK7完全失活所需的最短时间。用THZ1、THZ1-R或DMSO处理细胞0-6小时,以评估时间对THZ1介导的RNAPII CTD磷酸化抑制的影响。对于后续实验,除非另有说明,否则用上述时间过程实验确定的化合物处理细胞4小时。对于抑制剂洗脱实验(图2e,f;扩展数据图5),用THZ1、THZ1-R或DMSO处理细胞4小时。随后去除含有抑制剂的培养基以有效“洗脱”化合物,并允许细胞在没有抑制剂的情况下生长。对于每个实验,检测裂解物的RNAPII CTD磷酸化和其他特定蛋白质。 |
| 细胞实验 |
高通量细胞系平板活性测定[1]
将细胞接种在384孔微孔板中,在含有5%FBS和青霉素/链霉抗生物素的培养基中以约15%的融合率接种。用THZ1或DMSO处理细胞72小时,并用刃天青测定细胞存活率。 细胞增殖试验[2] 在病毒感染和嘌呤霉素选择后,将细胞接种在1ml培养基中的12孔板(密度为5×103)中。14天后,细胞用1%甲醛固定15分钟,用结晶紫(0.05%,wt/vol)染色15分钟,这是一种染色质结合细胞化学染色。这些板在大量去离子水中广泛清洗,在滤纸上倒置干燥,并用爱普生扫描仪成像。 对于96孔板中的3天细胞增殖试验,细胞以每孔6000至10000个细胞的密度铺板,并在第二天用不同浓度的THZ1或YKL-116处理。孵育72小时后,将CellTiter-Glo试剂直接加入细胞中,并在平板阅读器上读取发光信号。 |
| 参考文献 | |
| 其他信息 |
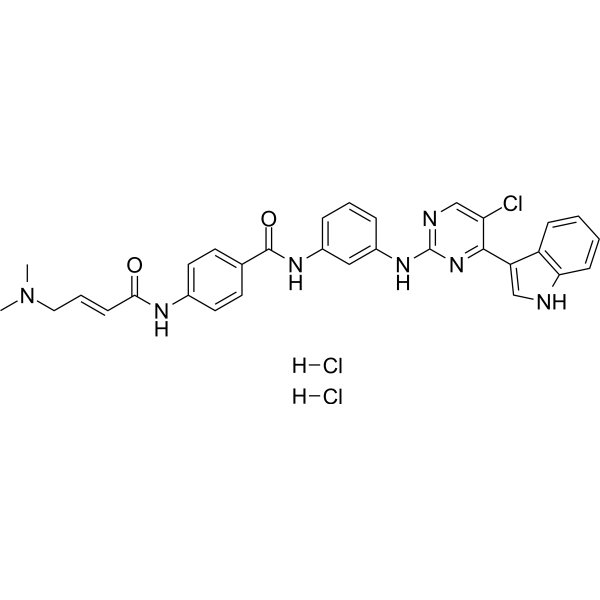
THZ1 is a member of the class of indoles that is 1H-indole substituted by a 5-chloro-2-[3-(4-{[(2E)-4-(dimethylamino)but-2-enoyl]amino}benzamido)anilino]pyrimidin-4-yl group at position 3. It is a selective and potent covalent inhibitor of CDK7 that exhibits anti-proliferative effects in cancer cell lines. It has a role as an EC 2.7.11.22 (cyclin-dependent kinase) inhibitor and an antineoplastic agent. It is a member of indoles, an aminopyrimidine, a member of benzamides, an organochlorine compound, an enamide and a secondary carboxamide.
Tumour oncogenes include transcription factors that co-opt the general transcriptional machinery to sustain the oncogenic state, but direct pharmacological inhibition of transcription factors has so far proven difficult. However, the transcriptional machinery contains various enzymatic cofactors that can be targeted for the development of new therapeutic candidates, including cyclin-dependent kinases (CDKs). Here we present the discovery and characterization of a covalent CDK7 inhibitor, THZ1, which has the unprecedented ability to target a remote cysteine residue located outside of the canonical kinase domain, providing an unanticipated means of achieving selectivity for CDK7. Cancer cell-line profiling indicates that a subset of cancer cell lines, including human T-cell acute lymphoblastic leukaemia (T-ALL), have exceptional sensitivity to THZ1. Genome-wide analysis in Jurkat T-ALL cells shows that THZ1 disproportionally affects transcription of RUNX1 and suggests that sensitivity to THZ1 may be due to vulnerability conferred by the RUNX1 super-enhancer and the key role of RUNX1 in the core transcriptional regulatory circuitry of these tumour cells. Pharmacological modulation of CDK7 kinase activity may thus provide an approach to identify and treat tumour types that are dependent on transcription for maintenance of the oncogenic state.[1] High-grade serous ovarian cancer is characterized by extensive copy number alterations, among which the amplification of MYC oncogene occurs in nearly half of tumors. We demonstrate that ovarian cancer cells highly depend on MYC for maintaining their oncogenic growth, indicating MYC as a therapeutic target for this difficult-to-treat malignancy. However, targeting MYC directly has proven difficult. We screen small molecules targeting transcriptional and epigenetic regulation, and find that THZ1 - a chemical inhibiting CDK7, CDK12, and CDK13 - markedly downregulates MYC. Notably, abolishing MYC expression cannot be achieved by targeting CDK7 alone, but requires the combined inhibition of CDK7, CDK12, and CDK13. In 11 patient-derived xenografts models derived from heavily pre-treated ovarian cancer patients, administration of THZ1 induces significant tumor growth inhibition with concurrent abrogation of MYC expression. Our study indicates that targeting these transcriptional CDKs with agents such as THZ1 may be an effective approach for MYC-dependent ovarian malignancies.[2] Small cell lung cancer (SCLC) is an aggressive disease with high mortality, and the identification of effective pharmacological strategies to target SCLC biology represents an urgent need. Using a high-throughput cellular screen of a diverse chemical library, we observe that SCLC is sensitive to transcription-targeting drugs, in particular to THZ1, a recently identified covalent inhibitor of cyclin-dependent kinase 7. We find that expression of super-enhancer-associated transcription factor genes, including MYC family proto-oncogenes and neuroendocrine lineage-specific factors, is highly vulnerability to THZ1 treatment. We propose that downregulation of these transcription factors contributes, in part, to SCLC sensitivity to transcriptional inhibitors and that THZ1 represents a prototype drug for tailored SCLC therapy.[3] The MYC oncoproteins are thought to stimulate tumor cell growth and proliferation through amplification of gene transcription, a mechanism that has thwarted most efforts to inhibit MYC function as potential cancer therapy. Using a covalent inhibitor of cyclin-dependent kinase 7 (CDK7) to disrupt the transcription of amplified MYCN in neuroblastoma cells, we demonstrate downregulation of the oncoprotein with consequent massive suppression of MYCN-driven global transcriptional amplification. This response translated to significant tumor regression in a mouse model of high-risk neuroblastoma, without the introduction of systemic toxicity. The striking treatment selectivity of MYCN-overexpressing cells correlated with preferential downregulation of super-enhancer-associated genes, including MYCN and other known oncogenic drivers in neuroblastoma. These results indicate that CDK7 inhibition, by selectively targeting the mechanisms that promote global transcriptional amplification in tumor cells, may be useful therapy for cancers that are driven by MYC family oncoproteins. Objectives: Oesophageal squamous cell carcinoma (OSCC) is an aggressive malignancy and the major histological subtype of oesophageal cancer. Although recent large-scale genomic analysis has improved the description of the genetic abnormalities of OSCC, few targetable genomic lesions have been identified, and no molecular therapy is available. This study aims to identify druggable candidates in this tumour. Design: High-throughput small-molecule inhibitor screening was performed to identify potent anti-OSCC compounds. Whole-transcriptome sequencing (RNA-Seq) and chromatin immunoprecipitation sequencing (ChIP-Seq) were conducted to decipher the mechanisms of action of CDK7 inhibition in OSCC. A variety of in vitro and in vivo cellular assays were performed to determine the effects of candidate genes on OSCC malignant phenotypes. [4] Results: The unbiased high-throughput small-molecule inhibitor screening led us to discover a highly potent anti-OSCC compound, THZ1, a specific CDK7 inhibitor. RNA-Seq revealed that low-dose THZ1 treatment caused selective inhibition of a number of oncogenic transcripts. Notably, further characterisation of the genomic features of these THZ1-sensitive transcripts demonstrated that they were frequently associated with super-enhancer (SE). Moreover, SE analysis alone uncovered many OSCC lineage-specific master regulators. Finally, integrative analysis of both THZ1-sensitive and SE-associated transcripts identified a number of novel OSCC oncogenes, including PAK4, RUNX1, DNAJB1, SREBF2 and YAP1, with PAK4 being a potential druggable kinase. Conclusions: Our integrative approaches led to a catalogue of SE-associated master regulators and oncogenic transcripts, which may significantly promote both the understanding of OSCC biology and the development of more innovative therapies.[5] |
| 分子式 |
C31H30CL3N7O2
|
|---|---|
| 分子量 |
638.97
|
| 精确质量 |
637.152
|
| CAS号 |
2095433-94-4
|
| PubChem CID |
129896819
|
| 外观&性状 |
Typically exists as solid at room temperature
|
| tPSA |
115
|
| 氢键供体(HBD)数目 |
6
|
| 氢键受体(HBA)数目 |
6
|
| 可旋转键数目(RBC) |
9
|
| 重原子数目 |
43
|
| 分子复杂度/Complexity |
896
|
| 定义原子立体中心数目 |
0
|
| SMILES |
CN(C)C/C=C/C(=O)NC1=CC=C(C=C1)C(=O)NC2=CC=CC(=C2)NC3=NC=C(C(=N3)C4=CNC5=CC=CC=C54)Cl.Cl.Cl
|
| InChi Key |
AJTGQOACYBCREM-QVLKBJGCSA-N
|
| InChi Code |
InChI=1S/C31H28ClN7O2.2ClH/c1-39(2)16-6-11-28(40)35-21-14-12-20(13-15-21)30(41)36-22-7-5-8-23(17-22)37-31-34-19-26(32)29(38-31)25-18-33-27-10-4-3-9-24(25)27;;/h3-15,17-19,33H,16H2,1-2H3,(H,35,40)(H,36,41)(H,34,37,38);2*1H/b11-6+;;
|
| 化学名 |
N-[3-[[5-chloro-4-(1H-indol-3-yl)pyrimidin-2-yl]amino]phenyl]-4-[[(E)-4-(dimethylamino)but-2-enoyl]amino]benzamide;dihydrochloride
|
| 别名 |
THZ1 Dihydrochloride; 2095433-94-4; THZ1 2HCl; (E/Z)-THZ1 (dihydrochloride); N-[3-[[5-chloro-4-(1H-indol-3-yl)pyrimidin-2-yl]amino]phenyl]-4-[[(E)-4-(dimethylamino)but-2-enoyl]amino]benzamide;dihydrochloride; THZ1Dihydrochloride; 1604810-83-4(free base);
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
Typically soluble in DMSO (e.g. 10 mM)
|
|---|---|
| 溶解度 (体内实验) |
注意: 如下所列的是一些常用的体内动物实验溶解配方,主要用于溶解难溶或不溶于水的产品(水溶度<1 mg/mL)。 建议您先取少量样品进行尝试,如该配方可行,再根据实验需求增加样品量。
注射用配方
注射用配方1: DMSO : Tween 80: Saline = 10 : 5 : 85 (如: 100 μL DMSO → 50 μL Tween 80 → 850 μL Saline)(IP/IV/IM/SC等) *生理盐水/Saline的制备:将0.9g氯化钠/NaCl溶解在100 mL ddH ₂ O中,得到澄清溶液。 注射用配方 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (如: 100 μL DMSO → 400 μL PEG300 → 50 μL Tween 80 → 450 μL Saline) 注射用配方 3: DMSO : Corn oil = 10 : 90 (如: 100 μL DMSO → 900 μL Corn oil) 示例: 以注射用配方 3 (DMSO : Corn oil = 10 : 90) 为例说明, 如果要配制 1 mL 2.5 mg/mL的工作液, 您可以取 100 μL 25 mg/mL 澄清的 DMSO 储备液,加到 900 μL Corn oil/玉米油中, 混合均匀。 View More
注射用配方 4: DMSO : 20% SBE-β-CD in Saline = 10 : 90 [如:100 μL DMSO → 900 μL (20% SBE-β-CD in Saline)] 口服配方
口服配方 1: 悬浮于0.5% CMC Na (羧甲基纤维素钠) 口服配方 2: 悬浮于0.5% Carboxymethyl cellulose (羧甲基纤维素) 示例: 以口服配方 1 (悬浮于 0.5% CMC Na)为例说明, 如果要配制 100 mL 2.5 mg/mL 的工作液, 您可以先取0.5g CMC Na并将其溶解于100mL ddH2O中,得到0.5%CMC-Na澄清溶液;然后将250 mg待测化合物加到100 mL前述 0.5%CMC Na溶液中,得到悬浮液。 View More
口服配方 3: 溶解于 PEG400 (聚乙二醇400) 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.5650 mL | 7.8251 mL | 15.6502 mL | |
| 5 mM | 0.3130 mL | 1.5650 mL | 3.1300 mL | |
| 10 mM | 0.1565 mL | 0.7825 mL | 1.5650 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
 CDK4 degrader 1
CDK4 degrader 1
 (+)-Atuveciclib
(+)-Atuveciclib
 EF-4-177
EF-4-177
 CDK8-IN-15
CDK8-IN-15
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1





























