
| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 5mg |
|
||
| 10mg |
|
||
| 50mg |
|
||
| Other Sizes |
|

| 靶点 |
p2x1 Receptor; P2X3 Receptor; P2X4 Receptor; P2X7 Receptor
|
|---|---|
| 体外研究 (In Vitro) |
BzATP(10-1000 μM;24 小时)刺激 U87 和 U251 胶质瘤细胞迁移和增殖 [3]。在人神经胶质瘤细胞中,BzATP(100μM;6-48小时)促进P2X7R蛋白的产生[3]。
|
| 体内研究 (In Vivo) |
与对照组和假手术组相比,盲肠结扎穿刺 (CLP) 诱导后,BzATP (5 mg/kg) 显着增加结肠中 P2X7R 的表达 [4]。
|
| 酶活实验 |
体外激活大鼠P2X3和P2X2/3受体[https://pubmed.ncbi.nlm.nih.gov/11156585/]
重组大鼠P2X3和大鼠P2X2/3受体cDNA与先前发表的用于大鼠同源和异源P2X3受体药理学体外表征的序列相同(Bianchi等人,1999)。使用标准脂质介导的转染方法构建稳定表达大鼠P2X3或大鼠P2X2/3受体的1321N1人星形细胞瘤细胞。所有细胞系均保存在含有10%FBS和抗生素的D-MEM中,如下所示:300μg ml-1 G418用于含P2X3的大鼠细胞;75μg ml-1潮霉素和150μg ml-1G418用于含P2X2/3的大鼠细胞。细胞在37°C的含5%二氧化碳的加湿气氛中生长。 P2X受体功能基于激动剂介导的细胞质Ca2+浓度的增加来确定,如前所述(Bianchi等人,1999)。BzATP(10μM)和α,β-meATP(10μM)分别用于激活大鼠P2X3和P2X2/3受体。简言之,使用荧光成像平板读取器(FLIPR),使用荧光Ca2+螯合染料(Fluo-4)作为96细胞形式的细胞内Ca2+的相对水平的指示剂。细胞在96孔黑壁组织培养板中生长至融合,并在23°C下在D-PBS中装载乙酰氧基甲酯(AM)形式的Fluo-4(1μM)1-2小时。在每次实验运行中,以1-5s的间隔收集荧光数据。使用GraphPad Prism中的四参数logistic Hill方程分析浓度响应数据。https://pubmed.ncbi.nlm.nih.gov/11156585/ |
| 细胞实验 |
细胞增殖测定[3]
细胞类型: U87 和 U251 神经胶质瘤细胞 测试浓度: 5、10、50、100、500 和 1000 μM 孵育时间:2、6、12、24、48和72小时 实验结果:U87和U251神经胶质瘤细胞系的增殖情况分别在 10-1000 μM 和 100-1000 μM 存在时显着增加。 U87和U251细胞系的细胞增殖峰值均在100μM处。 U87 和 U251 细胞系的最佳孵育时间均为 24 小时。 蛋白质印迹分析[3] 细胞类型: U87 和 U251 神经胶质瘤细胞 测试浓度: 100 μM 孵化持续时间:6-48 小时 实验结果:诱导 P2X7R 上调。 |
| 动物实验 |
Animal/Disease Models: Male 2-month-old C57BL/6 mice (each weighing between 20 and 25 g)[4]
Doses: 5 mg/kg Route of Administration: Injected through the intraperitoneal (ip)route Experimental Results: At 48 hrs (hours), mice in the treated group and The control group demonstrated mortalities of 91% and 86%, respectively. |
| 参考文献 |
|
| 其他信息 |
ATP functions as a fast neurotransmitter through the specific activation of a family of ligand-gated ion channels termed P2X receptors. In this report, six distinct recombinant P2X receptor subtypes were pharmacologically characterized in a heterologous expression system devoid of endogenous P2 receptor activity. cDNAs encoding four human P2X receptor subtypes (hP2X1, hP2X3, hP2X4, and hP2X7), and two rat P2X receptor subtypes (rP2X2 and rP2X3), were stably expressed in 1321N1 human astrocytoma cells. Furthermore, the rP2X2 and rP2X3 receptor subtypes were co-expressed in these same cells to form heteromultimeric receptors. Pharmacological profiles were determined for each receptor subtype, based on the activity of putative P2 ligands to stimulate Ca2+ influx. The observed potency and kinetics of each response was receptor subtype-specific and correlated with their respective electrophysiological properties. Each receptor subtype exhibited a distinct pharmacological profile, based on its respective sensitivity to nucleotide analogs, diadenosine polyphosphates and putative P2 receptor antagonists. Alphabeta-methylene ATP (alphabeta-meATP), a putative P2X receptor-selective agonist, was found to exhibit potent agonist activity only at the hP2X1, hP2X3 and rP2X3 receptor subtypes. Benzoylbenzoic ATP (BzATP, 2' and 3' mixed isomers), which has been reported to act as a P2X7 receptor-selective agonist, was least active at the rat and human P2X7 receptors, but was a potent (nM) agonist at hP2X1, rP2X3 and hP2X3 receptors. These data comprise a systematic examination of the functional pharmacology of P2X receptor activation. [1]
Agonist properties of the P2X7 receptor (P2X7R) differ strikingly from other P2X receptors in two main ways: high concentrations of ATP (> 100 microM) are required to activate the receptor, and the ATP analog 2',3'-O-(4-benzoyl-benzoyl)ATP (BzATP) is both more potent than ATP and evokes a higher maximum current. However, there are striking species differences in these properties. We sought to exploit the large differences in ATP and BzATP responses between rat and mouse P2X7R to delineate regions or specific residues that may be responsible for the unique actions of these agonists at the P2X7R. We measured membrane currents in response to ATP and BzATP at wild-type rat and mouse P2X7R, at chimeric P2X7Rs, and at mouse P2X7Rs bearing point mutations. Wild-type rat P2X7R was 10 times more sensitive to ATP and 100 times more sensitive to BzATP than wild-type mouse P2X7R. We found that agonist EC50 values were determined solely by the ectodomain of the P2X7R. Two segments (residues 115-136 and 282-288), when transposed together, converted mouse sensitivities to those of rat. Point mutations through these regions revealed a single residue, asparagine284, in the rat P2X7R that fully accounted for the 10-fold difference in ATP sensitivity, whereas the 100-fold difference in BzATP sensitivity required the transfer of both Lys127 and Asn284 from rat to mouse. Thus, single amino acid differences between species can account for large changes in agonist effectiveness and differentiate between the two widely used agonists at P2X7 receptors.[2] Previous studies have demonstrated that activation of P2X7 receptors (P2X7R) results in the proliferation and migration of some types of tumor. Here, we asked whether and how the activated P2X7R contribute to proliferation and migration of human glioma cells. Results showed that the number of P2X7R positive cells was increasing with grade of tumor. In U87 and U251 human glioma cell lines, both expressed P2X7R and the expression was enhanced by 3'-O-(4-benzoylbenzoyl) ATP (BzATP), the agonist of P2X7R, and siRNA. Our results also showed that 10 μM BzATP was sufficient to induce the proliferation of glioma cell significantly, while the cell proliferation reached the peak with 100 μM BzATP. Also, the migration of U87 and U251 cells was significantly increased upon BzATP treatment. However, the number of apoptotic cells of U87 and U251 was not significantly changed by BzATP. In addition, the expression of ERK, p-ERK, and proliferating cell nuclear antigen (PCNA) protein was increased in BzATP-treated U87 and U251 glioma cells. PD98059, an inhibitor of the MEK/ERK pathway, blocked the increased proliferation and migration of glioma cells activated by BzATP. These results suggest that ERK pathway is involved in the proliferation and migration of glioma cells induced by P2X7R activation.[3] |
| 分子式 |
C24H24N5O15P3.C18H45N3
|
|---|---|
| 分子量 |
1018.97
|
| 外观&性状 |
White to off-white solid powder
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month 注意: 请将本产品存放在密封且受保护的环境中,避免吸湿/受潮。 |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
H2O :~50 mg/mL (~49.07 mM)
|
|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: 100 mg/mL (98.14 mM) in PBS (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液; 超声助溶。
请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 0.9814 mL | 4.9069 mL | 9.8138 mL | |
| 5 mM | 0.1963 mL | 0.9814 mL | 1.9628 mL | |
| 10 mM | 0.0981 mL | 0.4907 mL | 0.9814 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
 P2X3 antagonist 39
P2X3 antagonist 39
 HEI3090
HEI3090
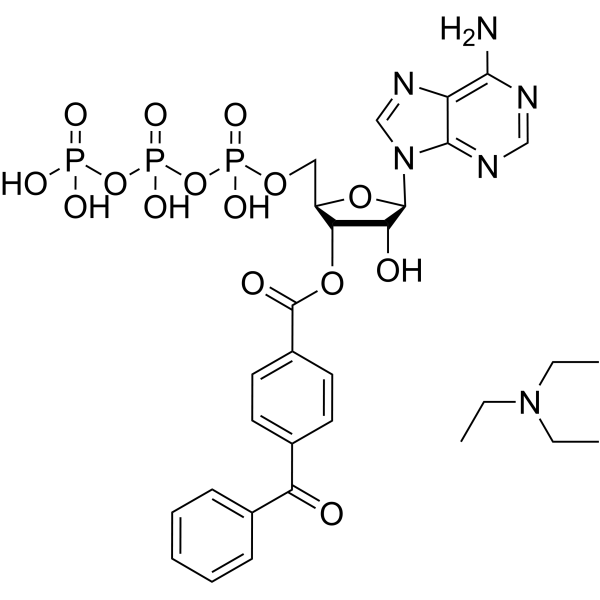 BzATP triethylammonium
BzATP triethylammonium
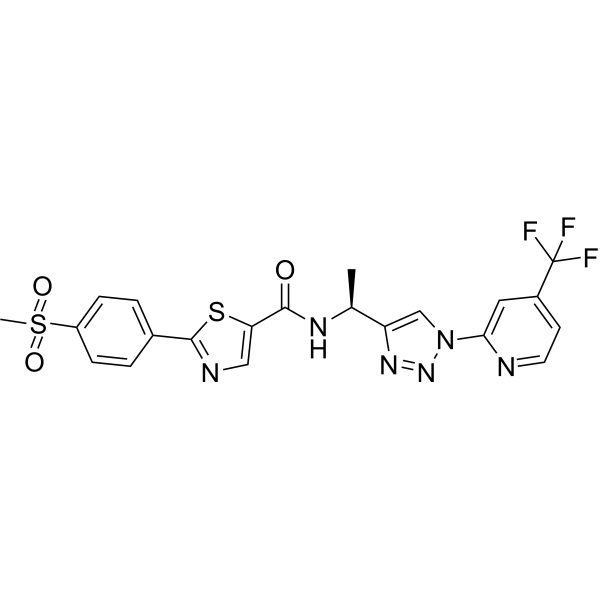 P2X7 receptor antagonist-5
P2X7 receptor antagonist-5
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1





























