| 规格 | 价格 | |
|---|---|---|
| 500mg | ||
| 1g | ||
| Other Sizes |

| 靶点 |
COX-2 (IC50 = 40 nM)
|
|---|---|
| 体外研究 (In Vitro) |
我们实验室最近的研究表明,底物类似物抑制剂(如5,8,11,14-二碳四萜酸)和非甾体抗炎药(如吲哚美辛和甲氯芬酸)中的羧酸部分衍生化会产生强效和选择性的环氧合酶-2 (COX-2)抑制剂(Kalgutkar等)。Proc。国家的。学会科学。美国,2000,97,925-930)。本文综述了芳基乙酸类非甾体抗炎药吲哚美辛转化为cox -2选择性抑制剂的结构-活性研究的细节。许多结构多样的吲哚美辛酯和酰胺在低纳摩尔范围内抑制纯化的人COX-2,其ICo5值在低纳摩尔范围内,但在高达66微米的浓度下不抑制绵羊COX-1的活性。吲哚美辛的一级和二级酰胺类似物比相应的叔酰胺更有效地抑制COX-2。吲哚美辛酯或酰胺中的4-氯苯甲酰被4-溴苄基官能团或氢取代,得到无活性化合物。同样,将酯和酰胺系列中吲哚环上的2-甲基与一个氢交换也会生成非活性化合物。抑制动力学表明,吲哚美辛酰胺表现为COX-2的缓慢,紧密结合抑制剂,并且选择性是时间依赖步骤的函数。将吲哚美辛转化为酯和酰胺衍生物为生成高选择性COX-2抑制剂和消除母体化合物的胃肠道副作用提供了一种简单的策略。[1]
|
| 酶活实验 |
酶学。[1]
如前所述,COX-1是从公羊精囊中纯化的蛋白质比活性为20 (μMO2/min)/mg,全蛋白含量为13.5%。ApoCOX-1的制备方法如前所述通过向测定混合物中加入血红蛋白来重组脱酶。利用pVL 1393表达载体在SF-9昆虫细胞中表达人COX-2,并通过离子交换和凝胶过滤层析纯化。通过7.5% SDS PAGE凝胶密度扫描,所有纯化蛋白的纯度均大于80%。 用薄层色谱(TLC)法测定绵羊COX-1和人COX-2抑制的时间和浓度依赖性[1] 用薄层色谱法测定了羊COX-1 (44 nM)和人COX-2 (66 nM)的环加氧酶活性。200 μL的反应混合物由血凝素重组蛋白在100 mM Tris-HCl、pH 8.0、500 μM苯酚和[1-14C]花生四烯酸(50 μM, ~ 55−57 mCi/mmol)中组成。对于时间依赖性的抑制实验,将血凝素重组的COX-1 (44 nM)或COX-2 (66 nM)与不同抑制剂浓度的DMSO在室温下预孵育20分钟,然后在37℃下加入[1-14C]花生四烯酸(50 μM) 30 s。在Et2O/CH3OH/1 M柠檬酸盐溶液中,pH 4.0(30:4:1),溶剂萃取终止反应。在2000g离心2min分离相,在TLC板上标记有机相。用EtOAc/CH2Cl2/ AcOH(75:25:1)溶液在4℃下显影。放射性标记的前列腺素产品用放射性扫描仪定量。在不同抑制剂浓度下观察到的总产物的百分比除以用DMSO预孵育相同时间的蛋白质样品观察到的产物的百分比。 |
| 细胞实验 |
活化RAW264.7中COX-2活性的抑制[1] < br >
在RAW264.7细胞中抑制COX-2的方法之前已经有过描述简单地说,用脂多糖(1 μg/mL)和γ-干扰素(10 U/mL)在无血清DMEM中活化细胞(6.2 × 106个细胞/T25瓶)7 h,然后用抑制剂(0 ~ 2 μM)在37℃下处理30 min。在37℃下,加入[1-14C]花生四烯酸(20 μM) 15 min,测定外源性花生四烯酸代谢。IC50值是两个独立测定值的平均值。
|
| 参考文献 |
[1]. Ester and amide derivatives of the nonsteroidal antiinflammatory drug, indomethacin, as selective cyclooxygenase-2 inhibitors. J Med Chem. 2000 Jul 27;43(15):2860-70.
|
| 其他信息 |
The present report describes our initial SAR studies of the conversion of indomethacin into COX-2-selective inhibitors using a biochemically based strategy that we recently reported. Neutral derivatives of NSAIDs such as indomethacin and meclofenamic acid retain the ability of the parent compounds to bind tightly to COX-2, but they do not bind to COX-1. Therefore, the conversion of these carboxylic acid-containing NSAIDs into ester or amide derivatives generates derivatives that are potent and highly selective COX-2 inhibitors. The potency of some of the derivatives reported in Tables 1−5 is underscored by the fact that the lowest IC50 values achieved against COX-2 are comparable to the enzyme concentration used in the assay (routinely 66 nM). The selectivity for COX-2 exhibited by the some of the most potent inhibitors is actually underestimated because many of the compounds assayed exhibited no inhibition of COX-1 at the highest inhibitor concentrations tested (66 μM). Thus, our relatively straightforward strategy produces excellent COX-2 inhibitors.[1]
Despite the fact that many of the esters and amides exhibited high COX-2 selectivity, there were notable exceptions, particularly in the ester series. For example, the 4-thiomethoxyphenyl ester of indomethacin was only 10-fold selective for COX-2, whereas the 2-thiomethoxyphenyl ester was greater than 1100-fold selective. Interestingly, the 4-methoxyphenyl ester was greater than 1700-fold selective, and the 4-thiomethoxyphenyl amide of indomethacin was greater than 550-fold selective. Likwise, the 4-fluorophenyl and 3-pyridyl esters exhibited only 40- and 50-fold selectivity, respectively, whereas the analogous amides were greater than 1000- and 1300-fold selective.[1] A striking observation from the present study is the breadth of substitutions tolerated in the ester or amide functionality that yield COX-2 inhibitors. Relatively large alkyl, aryl, aralkyl, and heterocyclic esters or amides of indomethacin exhibit high potency and selectivity. This is consistent with the model we proposed for the binding of these esters or amides to COX-2. The indomethacin moiety is located in the cyclooxygenase active site, but the ester and amide groups project through the constriction at the base of the active site and into a wide “lobby” in the membrane-binding domain (Figure 2). This model is consistent with the crystal structure of a complex of COX-2 with a carboxyl chain-extended analogue of zomepirac. A comparable lobby is present in COX-1, but there are a number of conserved sequence changes in this region between the proteins that may contribute to the COX-2 selectivity exhibited by ester and amide inhibitors.[1] Primary and secondary amide derivatives of indomethacin were potent inhibitors of COX-2, whereas tertiary amide derivatives were not. Several dialkyl amides, cycloalkyl amides, or pyridyl derivatives were synthesized and did not inhibit COX-2 under our standard assay conditions. This suggests that hydrogen bonding from the amide nitrogen to a protein acceptor is an important determinant of binding. Possible hydrogen bond acceptors near the carboxylate-binding region of COX-2 include Tyr355 and Glu524 (Figure 2). Mutation of either of these residues to Phe or Ala, respectively, renders the mutant proteins resistant to the inhibitory effects of indomethacin amides. Substitution of the p-chlorobenzoyl group of indomethacin with a p-bromobenzyl group generates a molecule, 77, that is a reasonably effective COX-2 inhibitor (IC50 = 2.5 μM, selectivity > 30 in our hands). We anticipated that conversion of this compound to esters or amides would generate a family of highly selective COX-2 inhibitors, but this was not the case. Neither the phenethyl ester 76 nor the phenethyl amide 81 exhibited any COX-2 inhibitory activity. The analogous derivatives of indomethacin, compounds 22 and 40, were highly selective inhibitors so it was quite surprising that the derivatives of 77 were not. This suggests that not all carboxylate-containing NSAIDs or related compounds will be converted into COX-2 inhibitors by esterification or amidation.[1] Perhaps the most surprising result from our SAR analysis was the complete absence of inhibitory activity of indomethacin esters or amides that lacked a methyl group at the 2-position of the indole ring (Table 5). This finding uncovers what may be a previously unrecognized determinant of COX inhibitory activity. Clearly, neither the 2-desmethylphenethyl ester nor the 2-desmethylphenethyl amide were active against COX-2. Since esters or amides of indomethacin are not time-dependent inhibitors of COX-1,38 it is not possible to speculate whether the 2-methyl group of nonselective inhibitors is also important for inhibition of COX-1. It will be interesting to prepare 2-desmethylindomethacin to explore this possibility.[1] Some of the molecules described herein (e.g., 40 and 57) have been found to exhibit antiinflammatory activity in the carageenan-induced foot pad edema assay following oral administration. The time dependence of antiiflammatory activity and in vitro analysis of metabolites generated by liver microsomes suggests that the antiinflammatory activity is due to the parent compound and not due to hydrolysis to indomethacin. This observation, coupled with the structural flexibility revealed in the present study, suggests that conversion of NSAIDs to amide derivatives may be a very useful approach for the generation of novel and efficacious COX-2 inhibitors. The ease of amide synthesis by combinatorial approaches should make it easy to construct highly diverse libraries to maximize not only COX-2 inhibitory activity but also physical properties, distribution, metabolic profile, etc. Thus, our biochemically based method for generating COX-2-selective inhibitors is a promising new strategy for the generation of antiinflammatory, analgesic, and antiangiogenic agents. |
| 分子式 |
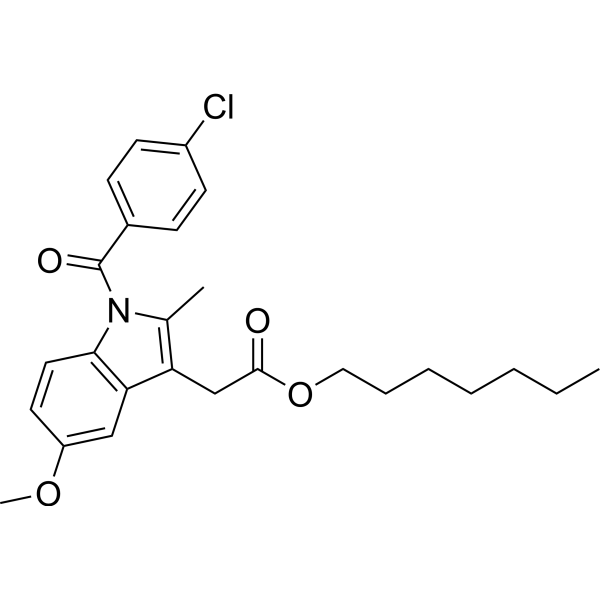
C26H30CLNO4
|
|---|---|
| 分子量 |
455.97
|
| 精确质量 |
455.186
|
| 元素分析 |
C, 68.49; H, 6.63; Cl, 7.77; N, 3.07; O, 14.03
|
| CAS号 |
282728-47-6
|
| PubChem CID |
10389320
|
| 外观&性状 |
Typically exists as solid at room temperature
|
| 密度 |
1.2±0.1 g/cm3
|
| 沸点 |
533.6±50.0 °C at 760 mmHg
|
| 闪点 |
276.5±30.1 °C
|
| 蒸汽压 |
0.0±1.4 mmHg at 25°C
|
| 折射率 |
1.565
|
| LogP |
6.75
|
| tPSA |
57.53
|
| SMILES |
CCCCCCCOC(=O)CC1=C(C)N(C2=C1C=C(C=C2)OC)C(=O)C3=CC=C(C=C3)Cl
|
| InChi Key |
PYBCHCVNKGZCOH-UHFFFAOYSA-N
|
| InChi Code |
InChI=1S/C26H30ClNO4/c1-4-5-6-7-8-15-32-25(29)17-22-18(2)28(24-14-13-21(31-3)16-23(22)24)26(30)19-9-11-20(27)12-10-19/h9-14,16H,4-8,15,17H2,1-3H3
|
| 化学名 |
heptyl 2-[1-(4-chlorobenzoyl)-5-methoxy-2-methylindol-3-yl]acetate
|
| 别名 |
Indomethacin heptyl ester; 282728-47-6; INDOMETHACIN ESTER, N-HEPTYL-; heptyl 2-[1-(4-chlorobenzoyl)-5-methoxy-2-methylindol-3-yl]acetate; CHEMBL330194; CHEBI:184056; HMS3649K19; BDBM50090775;
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples
|
|---|---|
| 溶解度 (体外实验) |
注意: 如下所列的是一些常用的体内动物实验溶解配方,主要用于溶解难溶或不溶于水的产品(水溶度<1 mg/mL)。 建议您先取少量样品进行尝试,如该配方可行,再根据实验需求增加样品量。
注射用配方
注射用配方1: DMSO : Tween 80: Saline = 10 : 5 : 85 (如: 100 μL DMSO → 50 μL Tween 80 → 850 μL Saline)(IP/IV/IM/SC等) *生理盐水/Saline的制备:将0.9g氯化钠/NaCl溶解在100 mL ddH ₂ O中,得到澄清溶液。 注射用配方 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (如: 100 μL DMSO → 400 μL PEG300 → 50 μL Tween 80 → 450 μL Saline) 注射用配方 3: DMSO : Corn oil = 10 : 90 (如: 100 μL DMSO → 900 μL Corn oil) 示例: 以注射用配方 3 (DMSO : Corn oil = 10 : 90) 为例说明, 如果要配制 1 mL 2.5 mg/mL的工作液, 您可以取 100 μL 25 mg/mL 澄清的 DMSO 储备液,加到 900 μL Corn oil/玉米油中, 混合均匀。 View More
注射用配方 4: DMSO : 20% SBE-β-CD in Saline = 10 : 90 [如:100 μL DMSO → 900 μL (20% SBE-β-CD in Saline)] 口服配方
口服配方 1: 悬浮于0.5% CMC Na (羧甲基纤维素钠) 口服配方 2: 悬浮于0.5% Carboxymethyl cellulose (羧甲基纤维素) 示例: 以口服配方 1 (悬浮于 0.5% CMC Na)为例说明, 如果要配制 100 mL 2.5 mg/mL 的工作液, 您可以先取0.5g CMC Na并将其溶解于100mL ddH2O中,得到0.5%CMC-Na澄清溶液;然后将250 mg待测化合物加到100 mL前述 0.5%CMC Na溶液中,得到悬浮液。 View More
口服配方 3: 溶解于 PEG400 (聚乙二醇400) 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.1931 mL | 10.9656 mL | 21.9313 mL | |
| 5 mM | 0.4386 mL | 2.1931 mL | 4.3863 mL | |
| 10 mM | 0.2193 mL | 1.0966 mL | 2.1931 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
 iNOS/COX-2-IN-1
iNOS/COX-2-IN-1
 Prostaglandin E2 Ethanolamide
Prostaglandin E2 Ethanolamide
 Isoscoparin-7-O-glucoside
Isoscoparin-7-O-glucoside
 COX-2-IN-41
COX-2-IN-41
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1
























