| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 5g |
|
||
| Other Sizes |
|

| 药代性质 (ADME/PK) |
Absorption, Distribution and Excretion
After intravenous administration, 95% of the dose is recovered in the urine after 24 hours. Oral administration in rats leads to 5.3% recovery in urine and 88.5% recovery in feces. Data regarding the volume of distribution of edetate disodium anhydrous is not readily available. The mean clearance of edetate in 1 month olds is 54.6mL/min/1.73m2. 2-17 year olds have a mean clearance of 113.9 ± 24.4 mL/min/1.73m2. After intravenous administration, the chelate formed is excreted in the urine with 50% appearing in 1 hour and over 95% in 24 hours. Disodium edentate ... /is/ poorly absorbed from the gastrointestinal tract and /is/ associated with few adverse effects when used as an excipient in pharmaceutical preparations. Twenty male Sprague-Dawley rats were divided into four groups of five animals each. Rats in group 1 received ip injections of (14)C Disodium EDTA, group 2 received this compound on depilated skin, rats in group 3 received this compound on depilated and abraded skin (abraded every 2 or 3 cm over treated area), and group 4 was the control group. The specific activity of the (14)C Disodium EDTA was 21.6 mCi/mM and it was dissolved in saline to yield a final solution of 50 pCi/mL. Animals that received ip injections got 0.5 mL of this solution, or 25 pCi of (14)C Disodium EDTA. Animals that had the compound applied to the skin received 25 pCi of (14)C Disodium EDTA in the form of an ointment (modulan, mineral oil, petrolatum, cetyl alcohol 35:21 :25:12) spread over an area of 50 sq cm spread over a sheet of thin polyethylene. This sheet was taped to the trunk of each animal. A collar was fixed around the neck of the rats. All animals were decapitated 24 hours after treatment. The tissue distribution (per 100 mg wet organ weight) of (14)C Disodium EDTA 24 hours after ip administration was as follows: liver 577+/- 13, small intestine 631 +/- 25, large intestine 696 +/- 19, and kidney 1964 +/- 220. Twenty-four hours after application on normal skin the tissue distribution was as follows: liver 6 +/- 4, small intestine 99 +/- 22, large intestine 107 +/- 24, and kidneys 29 +/- 12. Twenty-four hours after application on abraded skin the tissue distribution was as follows: liver 139 +/- 34, small intestine 214 +/- 76, large intestine 309 +/- 115, and kidneys 222 +/- 30. /Investigators/ reported that rats fed 0.5%, 1.0%, and 5.0% Disodium EDTA for 12 weeks excreted 82.2%, 44.5%, and 45.4%, respectively, of the ingested dose in the urine and feces. The feces contained 99.4%, 98.2%, and 97.5% of the excreted material and the urine contained 0.6%, 1.8%, and 2.5% of the material for the respective doses. For more Absorption, Distribution and Excretion (Complete) data for Disodium EDTA (7 total), please visit the HSDB record page. Metabolism / Metabolites Edetate is almost completely unmetabolized _in vivo_. Biological Half-Life After intravenous administration, the chelate formed is excreted in the urine with 50% appearing in 1 hour and over 95% in 24 hours. |
|---|---|
| 毒性/毒理 (Toxicokinetics/TK) |
Interactions
/Investigators/ reported that Disodium EDTA (10 mg/mL) increased the intestinal absorption of neutral, basic, and acidic compounds in the male Sprague-Dawley rat. The chelating agent increased the absorption of (14)C-mannitol and (14)C-inulin from <2% to 7%-b 1%, the absorption of (14)C-N-methyldecamethonium from 2%-3% to 11%-15%, and the absorption of sulfanilic acid from 11%-14% to 26%-32%. Plasma concentrations of the drugs were increased as much as five- or sixfold, compared to controls. Disodium EDTA at a concentration of 1% (w/v; 24 mM) increased the in situ drug absorption of acetazolamide from the small intestine of male Charles River rats when administered with 1% (w/v) reduced glutathione. Intestinal absorption was increased by 1.5 to 2 times; however, absorption from the stomach was not affected by treatment with EDTA and glutathione. The investigators suggested that Disodium EDTA altered the aqueous permeability of the intestinal epithelium by the chelation of magnesium and calcium ions, thereby separating the epithelial cells. Non-Human Toxicity Values LD50 Mouse oral 400 mg/kg LD50 Rat oral 3.7 g/kg LD50 Rabbit iv 47 mg/kg LD50 Rabbit oral 2300 mg/kg For more Non-Human Toxicity Values (Complete) data for Disodium EDTA (8 total), please visit the HSDB record page. |
| 参考文献 |
|
| 其他信息 |
Edetate disodium anhydrous is a polyvalent chelating agent used to treat hypercalcemia and digitalis toxicity associated ventricular arrhythmias.
Edetate Disodium is the disodium salt form of edetate, a heavy metal chelating agent with anti-hypercalcemic and anti-arrhythmic properties. Edetate, a heavy metal antagonist, chelates divalent and trivalent metals, forming soluble stable complexes which are readily excreted by the kidneys, thereby can be used to lower serum calcium concentrations. In addition, this agent exerts a negative inotropic effect on the heart through a transiently induced hypocalcemic state, thereby antagonizing the inotropic and chronotropic effects of digitalis glycosides on the ventricles of the heart. Upon ocular administration, edetate exerts its ophthalmic effect by chelating calcium to form soluble complexes, thereby removing corneal calcium deposits. A chelating agent that sequesters a variety of polyvalent cations such as CALCIUM. It is used in pharmaceutical manufacturing and as a food additive. See also: Edetic Acid (has active moiety); Aloe vera leaf; edetate disodium; glycerin; urea (component of). Drug Indication Edetate disodium is indicated for emergency treatment of hypercalcemia and digitalis toxicity associated ventricular arrhythmias. Mechanism of Action Edetate disodium anhydrous chelates divalent and trivalent ions such as magnesium, zinc, and calcium. The chelate is excreted in the urine, reducing concentrations of these ions in the blood. Edetate disodium injection forms chelates with the cations of calcium and many divalent and trivalent metals. Because of its affinity for calcium, edetate disodium will produce a lowering of the serum calcium level during intravenous infusion. Slow infusion over a protracted period may cause mobilization of extracirculatory calcium stores. Edetate disodium exerts a negative inotropic effect upon the heart. Edetate disodium likewise forms chelates with other polyvalent metals and produces increases in urinary excretion of magnesium, zinc and other trace elements. It does not form a chelate with potassium but may reduce the serum level and increase urinary loss of potassium. Therapeutic Uses Anticoagulants; Chelating Agents; Food Additives Endrate (Edetate Disodium Injection, USP) is indicated in selected patients for the emergency treatment of hypercalcemia and for the control of ventricular arrhythmias associated with digitalis toxicity. /Included in US product label/ Disodium edentate is also used therapeutically as an anticoagulant as it will chelate calcium and prevent the coagulation of blood in vitro. Concentrations of 0.1% w/v are used in small volumes for hematological testing and 0.3% w/v in transfusions. Disodium EDTA is used occasionally to terminate the effects of injected calcium, to antagonize digitalis toxicity, or to suppress tachyarrhythmias. /Former/ For more Therapeutic Uses (Complete) data for Disodium EDTA (8 total), please visit the HSDB record page. Drug Warnings /BOXED WARNING/ The use of this drug in any particular patient is recommended only when the severity of the clinical condition justifies the aggressive measures associated with this type of therapy. Clinical studies of edetate disodium did not include sufficient numbers of patients aged 65 and over to determine whether they respond differently from younger subjects. Other reported clinical experience has not identified differences in responses between elderly and younger patients. In general, dose selection for an elderly patient should be cautious, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy. Fatal medication errors have occurred that involve confusion between edetate calcium disodium (calcium EDTA) and edetate disodium (no longer commercially available in the US). Children and adults have mistakenly received edetate disodium instead of edetate calcium disodium; at least 5 deaths have occurred as a result of inadvertent administration of edetate disodium. Although both edetate calcium disodium and edetate disodium are heavy metal antagonists, the 2 drugs were originally approved by the US Food and Drug Administration (FDA) for different uses and have different effects; edetate disodium was formerly FDA approved for use in selected patients for the emergency treatment of hypercalcemia or for the control of ventricular arrhythmias associated with cardiac glycoside toxicity. Use of edetate disodium may result in a substantial, and sometimes fatal, decrease in serum calcium concentrations. In June 2008, FDA withdrew its prior approval for edetate disodium because of safety concerns following a review of the risk-benefit profile of the drug. FDA stated that it was not considering additional action regarding edetate calcium disodium at that time; most of the fatalities following administration of an EDTA drug have involved medication errors in which edetate disodium was administered instead of edetate calcium disodium. FDA has not received reports of any fatalities resulting from the administration of edetate calcium disodium that involve a medication error. Edetate Disodium Injection is contraindicated in anuric patients. It is not indicated for the treatment of generalized arteriosclerosis associated with advancing age. For more Drug Warnings (Complete) data for Disodium EDTA (22 total), please visit the HSDB record page. Pharmacodynamics Edetate disodium anhydrous is a polyvalent ion chelator that reduces blood concentrations of calcium or digitalis. It has a long duration of action as patients are generally given 1 daily dose. The therapeutic index is wide, as high doses are generally well tolerated. Patients should be counselled regarding the risk of postural hypotension, effects of myocardial contractility, hypokalemia, hypomagnesemia, and hypoglycemia. |
| 分子式 |
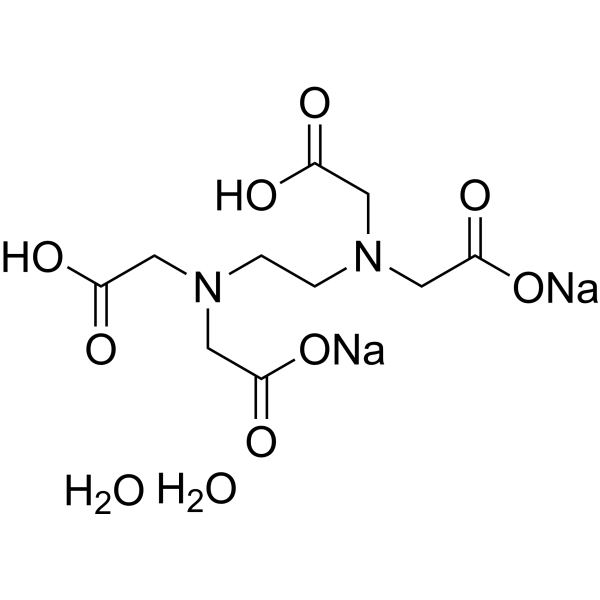
C10H18N2NA2O10
|
|---|---|
| 分子量 |
372.24
|
| 精确质量 |
372.075
|
| CAS号 |
6381-92-6
|
| 相关CAS号 |
Ethylenediaminetetraacetic acid;60-00-4
|
| PubChem CID |
636371
|
| 外观&性状 |
White to off-white solid powder
|
| 密度 |
1.01 g/mL at 25 °C
|
| 沸点 |
>100 °C
|
| 熔点 |
250 °C (dec.)(lit.)
|
| 闪点 |
325.2ºC
|
| 折射率 |
n20/D 1.363
|
| tPSA |
179.8
|
| 氢键供体(HBD)数目 |
4
|
| 氢键受体(HBA)数目 |
12
|
| 可旋转键数目(RBC) |
9
|
| 重原子数目 |
24
|
| 分子复杂度/Complexity |
336
|
| 定义原子立体中心数目 |
0
|
| InChi Key |
OVBJJZOQPCKUOR-UHFFFAOYSA-L
|
| InChi Code |
InChI=1S/C10H16N2O8.2Na.2H2O/c13-7(14)3-11(4-8(15)16)1-2-12(5-9(17)18)6-10(19)20;;;;/h1-6H2,(H,13,14)(H,15,16)(H,17,18)(H,19,20);;;2*1H2/q;2*+1;;/p-2
|
| 化学名 |
disodium;2-[2-[carboxylatomethyl(carboxymethyl)amino]ethyl-(carboxymethyl)amino]acetate;dihydrate
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month 注意: 请将本产品存放在密封且受保护的环境中,避免吸湿/受潮。 |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
H2O: 33.33 mg/mL (89.54 mM)
DMSO: < 1 mg/mL |
|---|---|
| 溶解度 (体内实验) |
注意: 如下所列的是一些常用的体内动物实验溶解配方,主要用于溶解难溶或不溶于水的产品(水溶度<1 mg/mL)。 建议您先取少量样品进行尝试,如该配方可行,再根据实验需求增加样品量。
注射用配方
注射用配方1: DMSO : Tween 80: Saline = 10 : 5 : 85 (如: 100 μL DMSO → 50 μL Tween 80 → 850 μL Saline)(IP/IV/IM/SC等) *生理盐水/Saline的制备:将0.9g氯化钠/NaCl溶解在100 mL ddH ₂ O中,得到澄清溶液。 注射用配方 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (如: 100 μL DMSO → 400 μL PEG300 → 50 μL Tween 80 → 450 μL Saline) 注射用配方 3: DMSO : Corn oil = 10 : 90 (如: 100 μL DMSO → 900 μL Corn oil) 示例: 以注射用配方 3 (DMSO : Corn oil = 10 : 90) 为例说明, 如果要配制 1 mL 2.5 mg/mL的工作液, 您可以取 100 μL 25 mg/mL 澄清的 DMSO 储备液,加到 900 μL Corn oil/玉米油中, 混合均匀。 View More
注射用配方 4: DMSO : 20% SBE-β-CD in Saline = 10 : 90 [如:100 μL DMSO → 900 μL (20% SBE-β-CD in Saline)] 口服配方
口服配方 1: 悬浮于0.5% CMC Na (羧甲基纤维素钠) 口服配方 2: 悬浮于0.5% Carboxymethyl cellulose (羧甲基纤维素) 示例: 以口服配方 1 (悬浮于 0.5% CMC Na)为例说明, 如果要配制 100 mL 2.5 mg/mL 的工作液, 您可以先取0.5g CMC Na并将其溶解于100mL ddH2O中,得到0.5%CMC-Na澄清溶液;然后将250 mg待测化合物加到100 mL前述 0.5%CMC Na溶液中,得到悬浮液。 View More
口服配方 3: 溶解于 PEG400 (聚乙二醇400) 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.6864 mL | 13.4322 mL | 26.8644 mL | |
| 5 mM | 0.5373 mL | 2.6864 mL | 5.3729 mL | |
| 10 mM | 0.2686 mL | 1.3432 mL | 2.6864 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
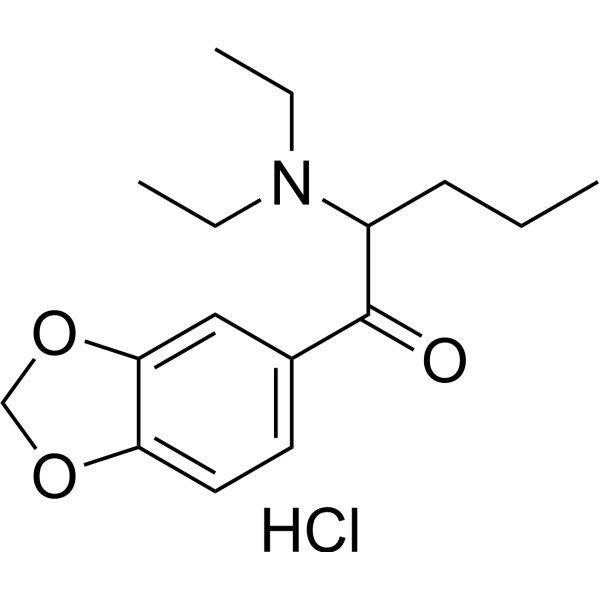 N,N-Diethylpentylone hydrochloride
N,N-Diethylpentylone hydrochloride
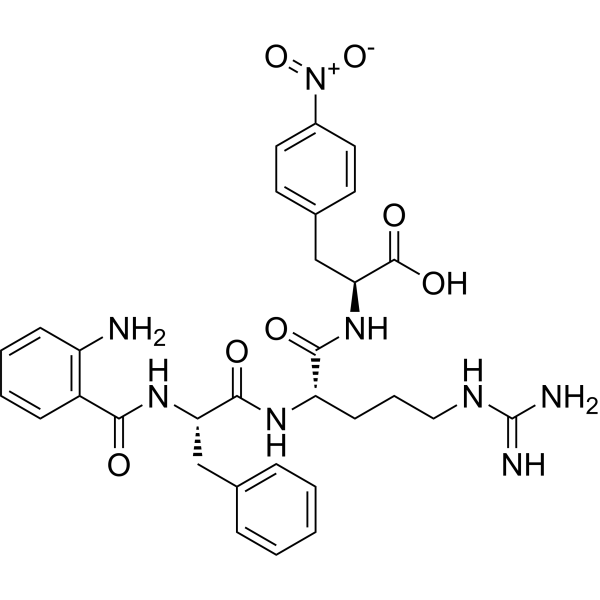 Abz-FRF(4NO2)
Abz-FRF(4NO2)
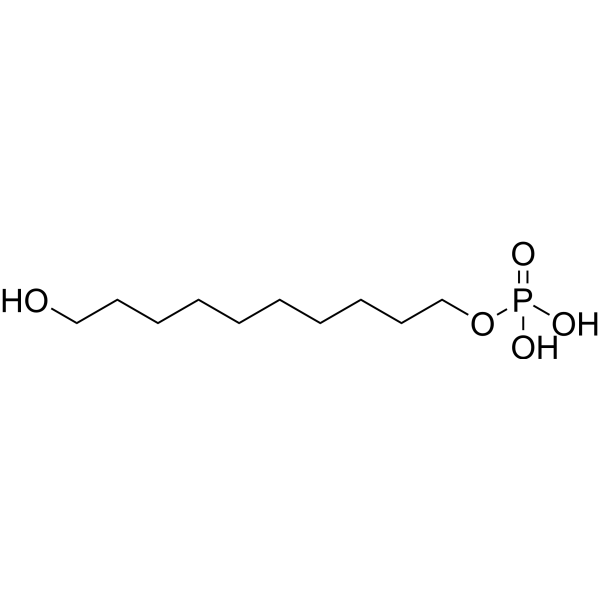 10-(Phosphonooxy)-1-decanole
10-(Phosphonooxy)-1-decanole
 Didocosanoin
Didocosanoin
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1





























