| 规格 | 价格 | |
|---|---|---|
| 500mg | ||
| 1g | ||
| Other Sizes |

| 靶点 |
hPPARγ (EC50 = 0.93 μM) mouse PPARγ (EC50 = 0.99 μM); hPPARδ (EC50 = 43 μM); hPPARα (EC50 = 100 μM); mouse PPARα (EC50 = 100 μM)
|
|---|---|
| 体外研究 (In Vitro) |
胰腺β细胞系T15可以通过完全防止AGEs诱导的β细胞坏死和caspase-3的升高来避免AGEs诱导的HIT损伤的细胞活力,其中吡格列酮钾(0.5或1μM,5天)效果最好有效的治疗可以预防这些事件的发生[2]。吡格列酮钾(1 μM,1 小时)可以降低 AGE 培养细胞中的 GSSG/GSH 比率,并增加低葡萄糖浓度引发的胰岛素分泌 [2]。
|
| 体内研究 (In Vivo) |
吡格列酮钾(口服强饲,10 或 30 mg/kg,每天一次,持续 14 天)可降低胰岛素抵抗和糖尿病,可能以脂质运载蛋白依赖性方式作用于肝脏,但不作用于骨骼肌[3]。吡格列酮钾(口服灌胃,10 mg/kg,每天一次,4周)可以显着降低体重(BW)、心脏肥大、血糖水平升高并改善相关的血脂异常[4]。
吡格列酮,每天口服一次,持续 14 天,剂量为 10 或 30 mg/kg,可改善糖尿病和胰岛素抵抗;这种作用在肝脏中可能依赖于脂质运载蛋白,但在骨骼肌中则不依赖[3]。吡格列酮(口服灌胃,10 mg/kg,每日一次,持续四周)可以缓解与之相关的血脂异常,提高血糖水平,并显着减轻体重(BW)和心脏肥厚[4]。 噻唑烷二酮已被证明可以上调白色脂肪组织中的脂联素表达和血浆脂联素水平,这些上调被认为是噻唑烷二酮类诱导改善与肥胖相关的胰岛素抵抗的主要机制。为了验证这一假设,我们生成了具有C57B/6背景的脂联素敲除(脂肪-/-)ob/ob小鼠。服用10mg/kg吡格列酮14天后,肥胖/肥胖小鼠的胰岛素抵抗和糖尿病明显改善,血清脂联素水平显著上调。肥胖/肥胖小鼠胰岛素抵抗的改善归因于肝脏中葡萄糖产量的减少和AMP活化蛋白激酶的增加,而不是骨骼肌中葡萄糖摄取的增加。相比之下,肥胖/肥胖/肥胖小鼠的胰岛素抵抗和糖尿病没有改善。服用30mg/kg吡格列酮14天后,肥胖/肥胖小鼠的胰岛素抵抗和糖尿病再次显著改善,这不仅归因于肝脏葡萄糖产量的减少,还归因于骨骼肌葡萄糖摄取的增加。有趣的是,肥胖/肥胖/肥胖小鼠也表现出胰岛素抵抗和糖尿病的显著改善,这归因于骨骼肌葡萄糖摄取的增加,而不是肝脏葡萄糖产量的减少。10mg/kg吡格列酮后,ob/ob和肥胖-/-ob/ob小鼠的血清游离脂肪酸和甘油三酯水平以及脂肪细胞大小没有变化,但30mg/kg吡格列酮类后显著降低到类似程度。此外,10mg/kg吡格列酮后,ob/ob和肥胖-/-ob/ob小鼠脂肪组织中TNFα和抵抗素的表达没有变化,但30mg/kg吡格列酮类后有所下降。因此,吡格列酮诱导的胰岛素抵抗和糖尿病的改善可能在肝脏中以脂联素依赖的方式发生,在骨骼肌中独立发生。[3] 吡格列酮已被证明对心血管结局有益。然而,人们对其对糖尿病肾病相关心脏重塑的影响知之甚少。因此,本研究旨在研究吡格列酮对糖尿病肾病大鼠模型心脏纤维化和肥大的影响。为此,将雄性Wistar白化大鼠随机分为4组(每组n=10):正常(n)组、糖尿病(D)组、接受等量赋形剂(0.5%羧甲基纤维素)的糖尿病肾病(DN)组和口服吡格列酮(10mg/kg/D)治疗4周的糖尿病肾病组。肾次全切除加链脲佐菌素(STZ)注射诱导糖尿病肾病。结果显示,DN大鼠的心脏组织中胶原纤维过度沉积,同时心肌细胞明显肥大。这与心脏转化生长因子β1(TGF-β1)基因的显著上调有关。此外,DN大鼠心脏中基质金属蛋白酶2(MMP-2)的基因表达降低,而金属蛋白酶组织抑制剂2(TIMP-2)的基因表达式升高。此外,在DN大鼠中观察到脂质过氧化和心肌损伤加剧,其血清肌酸激酶MB水平显著升高。服用吡格列酮后,所有这些异常都得到了改善。我们的研究结果表明,心脏TGF-β1基因的上调以及MMP-2和TIMP-2表达的不平衡与糖尿病肾病相关的心脏纤维化密切相关。吡格列酮可以通过抑制TGF-β1的基因表达和调节MMP-2/TIMP-2系统来改善心脏重塑[4]。 |
| 细胞实验 |
吡格列酮是一种抗糖尿病药物,可以保持胰腺β细胞质量并改善其功能。晚期糖基化终末产物(AGEs)与糖尿病并发症有关。我们之前已经证明,胰岛细胞系HIT-T15暴露于高浓度AGEs会显著降低细胞增殖和胰岛素分泌,并影响调节胰岛素基因转录的转录因子。本研究旨在探讨吡格列酮对AGEs培养的HIT-T15细胞功能和存活率的影响。HIT-T15细胞在单独存在AGEs或补充1μmol/l吡格列酮的情况下培养5天。然后测定细胞活力、胰岛素分泌和胰岛素含量、氧化还原平衡、AGE受体(RAGE)表达和NF-kB活化。结果表明,吡格列酮保护β细胞免受AGEs诱导的凋亡和坏死。此外,吡格列酮恢复了氧化还原平衡,提高了对低葡萄糖浓度的反应性。在AGEs培养物中添加吡格列酮可减弱NF-kB磷酸化,并阻止AGEs下调IkBα表达。这些发现表明,吡格列酮保护β细胞免受AGEs的危险影响[2]。
|
| 动物实验 |
Animal/Disease Models: ob/ob and adipo-/- ob/ob mice on C57Bl/6 background [3]
Doses: 10 or 30 mg/kg Route of Administration: po (oral gavage); one time/day; 14-day Experimental Results: ob/ Serum free fatty acid and triglyceride levels and adipocyte size were unchanged in ob and adipo-/- ob/ob C57BL/6 mice at the 10 mg/kg dose, but were Dramatically diminished to similar levels at the 10 mg/kg dose. degree. 30 mg/kg. It was also shown that the expression of TNFα and resistin in the adipose tissue of ob/ob and adipo-/- ob/ob mice was unchanged at the 10 mg/kg dose but diminished at the 30 mg/kg dose. Animal/Disease Models: Male Wistar albino rat [4] Doses: 10 mg/kg Route of Administration: po (oral gavage); one time/day; 4-week Experimental Results: Reduce elevated serum creatinine and creatine kinase MB (CK-MB) levels, TGF-β1 gene expression and regulates the expression of MMP-2/TIMP-2 system. |
| 药代性质 (ADME/PK) |
Absorption, Distribution and Excretion
Absorption Following oral administration of pioglitazone, peak serum concentrations are observed within 2 hours (Tmax) - food slightly delays the time to peak serum concentration, increasing Tmax to approximately 3-4 hours, but does not alter the extent of absorption. Steady-state concentrations of both parent drug and its primary active metabolites are achieved after 7 days of once-daily administration of pioglitazone. Cmax and AUC increase proportionately to administered doses. Route of Elimination Approximately 15-30% of orally administered pioglitazone is recovered in the urine. The bulk of its elimination, then, is presumed to be through the excretion of unchanged drug in the bile or as metabolites in the feces. Volume of Distribution The average apparent volume of distribution of pioglitazone is 0.63 ± 0.41 L/kg. Clearance The apparent clearance of orally administered pioglitazone is 5-7 L/h. There was no significant difference in the pharmacokinetic profile of pioglitazone in subjects with normal or with moderately impaired renal function. In patients with moderate and severe renal impairment, although mean serum concentrations of pioglitazone and its metabolites were increased, no dose adjustment is needed. After repeated oral doses of pioglitazone, mean AUC values were decreased in patients with severe renal impairment compared with healthy subjects with normal renal function for pioglitazone. Following oral administration, approximately 15% to 30% of the pioglitazone dose is recovered in the urine. Renal elimination of pioglitazone is negligible, and the drug is excreted primarily as metabolites and their conjugates. It is presumed that most of the oral dose is excreted into the bile either unchanged or as metabolites and eliminated in the feces. Pioglitazone is a thiazolidinedione insulin sensitizer that has shown efficacy in Type 2 diabetes and nonalcoholic fatty liver disease in humans. It may be useful for treatment of similar conditions in cats. The purpose of this study was to investigate the pharmacokinetics of pioglitazone in lean and obese cats, to provide a foundation for assessment of its effects on insulin sensitivity and lipid metabolism. Pioglitazone was administered intravenously (median 0.2 mg/kg) or orally (3 mg/kg) to 6 healthy lean (3.96 +/- 0.56 kg) and 6 obese (6.43 +/- 0.48 kg) cats, in a two by two Latin Square design with a 4-week washout period. Blood samples were collected over 24 hr, and pioglitazone concentrations were measured via a validated high-performance liquid chromatography assay. Pharmacokinetic parameters were determined using two-compartmental analysis for IV data and noncompartmental analysis for oral data. After oral administration, mean bioavailability was 55%, t(1/2) was 3.5 h, T(max) was 3.6 hr, C(max) was 2131 ng/mL, and AUC(0-8) was 15,56 ng/mL/hr. There were no statistically significant differences in pharmacokinetic parameters between lean and obese cats following either oral or intravenous administration. Systemic exposure to pioglitazone in cats after a 3 mg/kg oral dose approximates that observed in humans with therapeutic doses. PMID:22612529 The mean apparent volume of distribution (Vd/F) of pioglitazone following single-dose administration is 0.63 +/- 0.41 (mean +/- SD) L/kg of body weight. Pioglitazone is extensively protein bound (> 99%) in human serum, principally to serum albumin. Pioglitazone also binds to other serum proteins, but with lower affinity. M-III (keto derivative of pioglitazone) and M-IV (hydroxyl derivative of pioglitazone) are also extensively bound (> 98%) to serum albumin. View More
Metabolism / Metabolites Pioglitazone is extensively metabolized by both hydroxylation and oxidation - the resulting metabolites are also partly converted to glucuronide or sulfate conjugates. The pharmacologically active M-IV and M-III metabolites are the main metabolites found in human serum and their circulating concentrations are equal to, or greater than, those of the parent drug. The specific CYP isoenzymes involved in the metabolism of pioglitazone are CYP2C8 and, to a lesser degree, CYP3A4. There is also some evidence to suggest a contribution by extrahepatic CYP1A1. Isoforms of cytochrome P450 (CYP) are involved in the metabolism of pioglitazone, including CYP2C8 and, to a lesser degree, CYP3A4. CYP2C9 is not significantly involved in the elimination of pioglitazone. Pioglitazone is not a strong inducer of CYP3A4, and pioglitazone was not shown to induce CYPs. Pioglitazone is extensively metabolized by hydroxylation and oxidation; the metabolites also partly convert to glucuronide or sulfate conjugates. Metabolites M-III (keto derivative of pioglitazone) and M-IV (hydroxyl derivative of pioglitazone) are the major circulating active metabolites in humans. Pioglitazone has known human metabolites that include 2-[6-(2-{4-[(2,4-dioxo-1,3-thiazolidin-5-yl)methyl]phenoxy}ethyl)pyridin-3-yl]acetic acid, 5-[(4-{2-[5-(1-hydroxyethyl)pyridin-2-yl]ethoxy}phenyl)methyl]-1,3-thiazolidine-2,4-dione, and 5-({4-[2-(5-ethylpyridin-2-yl)-2-hydroxyethoxy]phenyl}methyl)-1,3-thiazolidine-2,4-dione. Biological Half-Life The mean serum half-life of pioglitazone and its metabolites (M-III and M-IV) range from 3-7 hours and 16-24 hours, respectively. The mean serum half-life of pioglitazone and its metabolites (M-III and M-IV) range from three to seven hours and 16 to 24 hours, respectively. |
| 毒性/毒理 (Toxicokinetics/TK) |
Toxicity Summary
IDENTIFICATION AND USE: Pioglitazone is a solid. It is used as hypoglycemic agent as an adjunct to diet and exercise for the management of type 2 diabetes mellitus. HUMAN STUDIES: Pioglitazone hydrochloride is a thiazolidinedione that depends on the presence of insulin for its mechanism of action. Pioglitazone hydrochloride decreases insulin resistance in the periphery and in the liver resulting in increased insulin-dependent glucose disposal and decreased hepatic glucose output. Pioglitazone is an agonist for peroxisome proliferator-activated receptor-gamma (PPARgamma). PPAR receptors are found in tissues important for insulin action such as adipose tissue, skeletal muscle, and liver. Activation of PPARgamma nuclear receptors modulates the transcription of a number of insulin responsive genes involved in the control of glucose and lipid metabolism. No evidence of hepatotoxicity has been noted with pioglitazone in clinical studies to date. However, hepatitis, liver function test abnormalities (such as elevations in hepatic enzymes to at least 3 times the upper limit of normal), mixed hepatocellular-cholestatic liver injury, and liver failure with or without fatalities have been reported during postmarketing experience with the drug. Thiazolidinediones, including pioglitazone hydrochloride, cause or exacerbate congestive heart failure in some patients. Pioglitazone-induced heart failure is known in patients with underlying heart disease, but is not well documented in patients with normal left ventricular function. It has been however reported that a patient developed congestive heart failure and pulmonary edema with normal left ventricular function within 1 year of starting pioglitazone therapy. Patients treated with pioglitazone have increased risk of bladder cancer compared to general population. There was also described an association of pioglitazone use with an increased risk of newly developed chronic kidney disease. ANIMAL STUDIES: Heart enlargement was seen in a 13-week study in monkeys at oral doses of 8.9 mg/kg and above, but not in a 52-week study at oral doses up to 32 mg/kg. Heart enlargement has been observed in mice (100 mg/kg), rats (4 mg/kg and above) and dogs (3 mg/kg) treated orally with pioglitazone hydrochloride. In a one-year rat study, drug-related early death due to apparent heart dysfunction occurred at an oral dose of 160 mg/kg/day. A two-year carcinogenicity study was conducted in male and female rats at oral doses up to 63 mg/kg. Drug-induced tumors were not observed in any organ except for the urinary bladder. A two-year carcinogenicity study was conducted in male and female mice at oral doses up to 100 mg/kg/day. No drug-induced tumors were observed in any organ. No adverse effects upon fertility were observed in male and female rats at oral doses up to 40 mg/kg pioglitazone hydrochloride daily prior to and throughout mating and gestation. Pioglitazone administered to pregnant rats during organogenesis did not cause adverse developmental effects at a dose of 20 mg/kg. When pregnant rats received pioglitazone during late gestation and lactation, delayed postnatal development, attributed to decreased body weight, occurred in offspring at maternal doses of 10 mg/kg and above. In pregnant rabbits administered pioglitazone during organogenesis, no adverse developmental effects were observed at 80 mg/kg, but reduced embryofetal viability at 160 mg/kg. Pioglitazone hydrochloride was not mutagenic in a battery of genetic toxicology studies, including the Ames bacterial assay, a mammalian cell forward gene mutation assay, an in vitro cytogenetics assay using CHL cells, an unscheduled DNA synthesis assay, and an in vivo micronucleus assay. Pioglitazone acts as an agonist at peroxisome proliferator activated receptors (PPAR) in target tissues for insulin action such as adipose tissue, skeletal muscle, and liver. Activation of PPAR-gamma receptors increases the transcription of insulin-responsive genes involved in the control of glucose production, transport, and utilization. In this way, pioglitazone both enhances tissue sensitivity to insulin and reduces hepatic gluconeogenesis. Thus, insulin resistance associated with type 2 diabetes mellitus is improved without an increase in insulin secretion by pancreatic β cells. View More
Hepatotoxicity
◉ Summary of Use during Lactation No information is available on the clinical use of pioglitazone during breastfeeding. Pioglitazone is over 99% protein bound in plasma, so it is unlikely to pass into breastmilk in clinically important amounts. However, an alternate drug may be preferred, especially while nursing a newborn or preterm infant. ◉ Effects in Breastfed Infants Relevant published information was not found as of the revision date. ◉ Effects on Lactation and Breastmilk Relevant published information was not found as of the revision date. Exposure Routes Following oral administration, in the fasting state, pioglitazone is first measurable in serum within 30 minutes, with peak concentrations observed within 2 hours. Food slightly delays the time to peak serum concentration to 3 to 4 hours, but does not alter the extent of absorption. Toxicity Data Hypogycemia; LD50=mg/kg (orally in rat) Interactions The aim was to investigate the effects of coadministration of the sodium glucose cotransporter 2 (SGLT2) inhibitor empagliflozin with the thiazolidinedione pioglitazone. In study 1, 20 healthy volunteers received 50 mg of empagliflozin alone for 5 days, followed by 50 mg of empagliflozin coadministered with 45 mg of pioglitazone for 7 days and 45 mg of pioglitazone alone for 7 days in 1 of 2 treatment sequences. In study 2, 20 volunteers received 45 mg of pioglitazone alone for 7 days and 10, 25, and 50 mg of empagliflozin for 9 days coadministered with 45 mg of pioglitazone for the first 7 days in 1 of 4 treatment sequences. Pioglitazone exposure (Cmax and AUC) increased when coadministered with empagliflozin versus monotherapy in study 1. The geometric mean ratio (GMR) for pioglitazone Cmax at steady state (Cmax,ss) and for AUC during the dosing interval at steady state (AUCt,ss) when coadministered with empagliflozin versus administration alone was 187.89% (95% CI, 166.35%-212.23%) and 157.97% (95% CI, 148.02%-168.58%), respectively. Because an increase in pioglitazone exposure was not expected, based on in vitro data, a second study was conducted with the empagliflozin doses tested in Phase III trials. In study 2, pioglitazone exposure decreased marginally when coadministered with empagliflozin. The GMR for pioglitazone Cmax,ss when coadministered with empagliflozin versus administration alone was 87.74% (95% CI, 73.88%-104.21%) with empagliflozin 10 mg, 90.23% (95% CI, 66.84%-121.82%) with empagliflozin 25 mg, and 89.85% (95% CI, 71.03%-113.66%) with empagliflozin 50 mg. The GMR for pioglitazone AUCt,ss when coadministered with empagliflozin versus administration alone was 90.01% (95% CI, 77.91%-103.99%) with empagliflozin 10 mg, 88.98% (95% CI, 72.69%-108.92%) with empagliflozin 25 mg, and 91.10% (95% CI, 77.40%-107.22%) with empagliflozin 50 mg. The effects of empagliflozin on pioglitazone exposure are not considered to be clinically relevant. Empagliflozin exposure was unaffected by coadministration with pioglitazone. Empagliflozin and pioglitazone were well tolerated when administered alone or in combination. In study 1, adverse events were reported in 1 of 19 participants on empagliflozin 50 mg alone, 4 of 20 on pioglitazone alone, and 5 of 18 on combination treatment. In study 2, adverse events were reported in 8 of 20 participants on pioglitazone alone, 10 of 18 when coadministered with empagliflozin 10 mg, 5 of 17 when coadministered with empagliflozin 25 mg, and 6 of 16 when coadministered with empagliflozin 50 mg. These results indicate that pioglitazone and empagliflozin can be coadministered without dose adjustments. PMID:26051874 The thiazolidinedione antidiabetic drug pioglitazone is metabolized mainly by cytochrome P450 (CYP) 2C8 and CYP3A4 in vitro. Our objective was to study the effects of gemfibrozil, itraconazole, and their combination on the pharmacokinetics of pioglitazone to determine the role of these enzymes in the fate of pioglitazone in humans. In a randomized, double-blind, 4-phase crossover study, 12 healthy volunteers took either 600 mg gemfibrozil or 100 mg itraconazole (first dose, 200 mg), both gemfibrozil and itraconazole, or placebo twice daily for 4 days. On day 3, they received a single dose of 15 mg pioglitazone. Plasma drug concentrations and the cumulative excretion of pioglitazone and its metabolites into urine were measured for up to 48 hours. RESULTS: Gemfibrozil alone raised the mean total area under the plasma concentration-time curve from time 0 to infinity [AUC(0-infinity)] of pioglitazone 3.2-fold (range, 2.3-fold to 6.5-fold; P < 0.001) and prolonged its elimination half-life (t (1/2) ) from 8.3 to 22.7 hours ( P < .001) but had no significant effect on its peak concentration (C max ) compared with placebo (control). Gemfibrozil increased the 48-hour excretion of pioglitazone into urine by 2.5-fold ( P < 0.001) and reduced the ratios of the active metabolites M-III and M-IV to pioglitazone in plasma and urine. Gemfibrozil decreased the area under the plasma concentration-time curve from time 0 to 48 hours [AUC(0-48)] of the metabolites M-III and M-IV by 42% ( P < 0.05) and 45% ( P < 0.001), respectively, but their total AUC(0-infinity) values were reduced by less or not at all. Itraconazole had no significant effect on the pharmacokinetics of pioglitazone and did not alter the effect of gemfibrozil on pioglitazone pharmacokinetics. The mean area under the concentration versus time curve to 49 hours [AUC(0-49)] of itraconazole was 46% lower ( P <0.001) during the gemfibrozil-itraconazole phase than during the itraconazole phase. Gemfibrozil elevates the plasma concentrations of pioglitazone, probably by inhibition of its CYP2C8-mediated metabolism. CYP2C8 appears to be of major importance and CYP3A4 of minor importance in pioglitazone metabolism in vivo in humans. Concomitant use of gemfibrozil with pioglitazone may increase the effects and risk of dose-related adverse effects of pioglitazone. However, studies in diabetic patients are needed to determine the clinical significance of the gemfibrozil-pioglitazone interaction. PMID:15900286 Domperidone (prokinetic agent) is frequently co-administered with pioglitazone (anitidiabetic) or ondansetron (antiemetic) in gastroparesis management. These drugs are metabolized via cytochome P-450 (CYP) 3A4, raising the possibility of interaction and adverse reactions. The concentration-dependent inhibitory effect of pioglitazone and ondansetron on domperidone hydroxylation was monitored in pooled human liver microsomes (HLM). Pioglitazone was further assessed as a mechanism-based inhibitor. Microsomal binding was evaluated in our assessment. In HLM, Vmax/Km estimates for monohydroxy domperidone formation decreased in presence of pioglitazone. Diagnostic plots indicated that pioglitazone inhibited domperidone in a partial mixed-type manner. The in vitro Ki was 1.52 uM. Predicted in vivo AUCi/AUC ratio was 1.98. Pioglitazone also exerted time-dependent inhibition on the metabolism of domperidone and the average remaining enzymatic activity decreased significantly upon preincubation with pioglitazone over 0-40 min. Diagnostic plots showed no inhibitory effect of ondansetron on domperidone hydroxylation. In conclusion, pioglitazone inhibited domperidone metabolism in vitro through different complex mechanisms. Our in vitro data predict that the co-administration of these drugs can potentially trigger an in vivo drug-drug interaction. PMID:24641107 /The objective of this study was/ to investigate potential drug-drug interactions between topiramate and metformin and pioglitazone at steady state. Two open-label studies were performed in healthy adult men and women. In Study 1, eligible participants were given metformin alone for 3 days (500 mg twice daily (BID)) followed by concomitant metformin and topiramate (titrated to 100 mg BID) from days 4 to 10. In Study 2, eligible participants were randomly assigned to treatment with pioglitazone 30 mg once daily (QD) alone for 8 days followed by concomitant pioglitazone and topiramate (titrated to 96 mg BID) from days 9 to 22 (Group 1) or to topiramate (titrated to 96 mg BID) alone for 11 days followed by concomitant pioglitazone 30 mg QD and topiramate 96 mg BID from days 12 to 22 (Group 2). An analysis of variance was used to evaluate differences in pharmacokinetics with and without concomitant treatment; 90% confidence intervals (CI) for the ratio of the geometric least squares mean (LSM) estimates for maximum plasma concentration (Cmax), area under concentration-time curve for dosing interval (AUC12 or AUC24), and oral clearance (CL/F) with and without concomitant treatment were used to assess a drug interaction. A comparison to historical data suggested a modest increase in topiramate oral clearance when given concomitantly with metformin. Coadministration with topiramate reduced metformin oral clearance at steady state, resulting in a modest increase in systemic metformin exposure. Geometric LSM ratios and 90% CI for metformin CL/F and AUC12 were 80% (75%, 85%) and 125% (117%, 134%), respectively. Pioglitazone had no effect on topiramate pharmacokinetics at steady state. Concomitant topiramate resulted in decreased systemic exposure to pioglitazone and its active metabolites, with geometric LSM ratios and 90% CI for AUC24 of 85.0% (75.7%, 95.6%) for pioglitazone, 40.5% (36.8%, 44.6%) for M-III, and 83.8% (76.1%, 91.2%) for M-IV, respectively. This effect appeared more pronounced in women than in men. Coadministration of topiramate with metformin or pioglitazone was generally well tolerated by healthy participants in these studies. A modest increase in metformin exposure and decrease in topiramate exposure was observed at steady state following coadministration of metformin 500 mg BID and topiramate 100mg BID. The clinical significance of the observed interaction is unclear but is not likely to require a dose adjustment of either agent. Pioglitazone 30 mg QD did not affect the pharmacokinetics of topiramate at steady state, while coadministration of topiramate 96 mg BID with pioglitazone decreased steady-state systemic exposure to pioglitazone, M-III, and M-IV. While the clinical consequence of this interaction is unknown, careful attention should be given to the routine monitoring for adequate glycemic control of patients receiving this concomitant therapy. Concomitant administration of topiramate with metformin or pioglitazone was generally well tolerated and no new safety concerns were observed. PMID:25219351 Protein Binding Pioglitazone is >99% protein-bound in human plasma - binding is primarily to albumin, although pioglitazone has been shown to bind other serum proteins with a lower affinity. The M-III and M-IV metabolites of pioglitazone are >98% protein-bound (also primarily to albumin). Ecological Information Environmental Fate / Exposure Summary Pioglitazone's production and administration as a medication may result in its release to the environment through various waste streams. If released to air, an estimated vapor pressure of 2.9X10-14 mm Hg at 25 °C indicates pioglitazone will exist solely in the particulate phase in the atmosphere. Particulate-phase pioglitazone will be removed from the atmosphere by wet and dry deposition. Pioglitazone contains pyridine- and anisole-like functional groups that do not absorb UV light at wavelengths >290 nm and, therefore, may be susceptible to direct photolysis by sunlight. If released to soil, pioglitazone is expected to be immobile based upon an estimated Koc of 9000. Volatilization from moist soil surfaces is not expected to be an important fate process based upon an estimated Henry's Law constant of 1.7X10-12 atm-cu m/mole. Pioglitazone is not expected to volatilize from dry soil surfaces based upon its vapor pressure. Biodegradation data ln soil or water where not available. If released into water, pioglitazone is expected to adsorb to suspended solids and sediment based upon the estimated Koc. Volatilization from water surfaces is not expected to be an important fate process based upon this compound's estimated Henry's Law constant. An estimated BCF of 190 suggests the potential for bioconcentration in aquatic organisms is high. Hydrolysis is not expected to be an important environmental fate process since this compound lacks functional groups that hydrolyze under environmental conditions (pH 5 to 9). Occupational exposure to pioglitazone may occur through inhalation and dermal contact with this compound at workplaces where pioglitazone is produced or used. The general public is not likely to be exposed to pioglitazone unless by direct medical treatment. (SRC) History and Incidents Rosiglitazone was approved by the U.S. Food and Drug Administration (FDA) for type 2 diabetes in 1999. The unique mechanism of action and low risk of hypoglycemia contributed to rapid market uptake of rosiglitazone, but safety concerns became more prominent in 2007. There were 5 major events on 4 calendar days in 2007 regarding safety concerns related to rosiglitazone in certain patients: (1) the May 21, 2007, online release of the rosiglitazone meta-analysis performed by Nissen and Wolski and the FDA safety warning on the same day; (2) the July 30, 2007, conclusion of an FDA advisory committee meeting that rosiglitazone increased cardiac ischemic risk; (3) the August 14, 2007, update of thiazolidinedione (TZD) labels with a black-box warning for heart failure; and (4) the November 14, 2007, update to the warnings and precautions section of the rosiglitazone label for coadministration of nitrate or insulin. /The purpose of this paper was/ to (1) describe TZD (rosiglitazone and pioglitazone) utilization trends from January 1, 2007, continuing through May 2008 amid public announcements of safety concerns and (2) determine the percentage of TZD users who had medical claims indicating increased cardiovascular (CV) risk before and after release (May 21, 2007) of the FDA safety warning and online release of the meta-analysis performed by Nissen and Wolski. A retrospective analysis of pharmacy claims was performed from 9 commercial plans with a combined 9 million eligible members, including a 1.4 million-member cohort from 1 of the plans for which medical claims data were available. We evaluated trends in TZD use for each month for the 17-month period from January 1, 2007, through May 31, 2008, including the percentage of TZD users at increased CV risk. In the trend analysis, for each calendar month of 2007, we calculated mean pharmacy claim counts per day per million members for each of the 2 TZD drugs and for a comparison drug, sitagliptin, a new oral hypoglycemic agent in a different class (dipeptidyl-peptidase-IV inhibitors). For the CV risk analysis, we used the database of integrated medical and pharmacy claims for the 1.4 million-member cohort to identify patients with a current days supply of a TZD on May 20, 2007, December 7, 2007, or May 20, 2008. The medical claims for all identified patients were queried back 2 years from May 20, 2007, December 7, 2007, or May 20, 2008, respectively. Rosiglitazone users at increased CV rsk were defined as those with a medical claim with a primary diagnosis for congestive heart failure (CHF; International Classification of Diseases, Ninth Revision, Clinical Modification [ICD-9-CM] codes 428.xx or 398.91), those with a current supply of nitrate or insulin therapy, or those with ischemic heart disease, including myocardial infarction (MI; ICD-9-CM codes 410.xx through 414.xx, or surgical procedure codes [36.0x through 36.3x for removal of obstruction and insertion of stents, bypass surgery, and revascularization] in the primary diagnosis field). Pioglitazone users at increased risk were identified from medical claims with a CHF diagnosis code. The average number of claims per day per million members in January 2007 was 97.3 for rosiglitazone and 107.2 for pioglitazone. The average number of claims for rosiglitazone per day per million members began to decrease in May 2007, falling to 41.0 in December 2007, for a total decrease of 58.6% from the February 2007 peak (99.1), and fell further to 31.8 in May 2008. Pioglitazone use increased 8.0% from January to June 2007 (107.2 to 115.8) and remained relatively flat through December 2007 (114.6) and through May 2008 (108.9). Sitagliptin claims increased 5-fold, at a consistent rate, from an average of 8.6 claims per day per million members in January 2007 to 43.4 in December 2007, and continued to increase to 48.7, in May 2008. Of the 5,117 rosiglitazone users on May 20, 2007, 1,296 (25.3%) were identified at increased CV risk versus 590 (22.5%) of 2,621 users on December 7, 2007 (P = 0.006), and 336 (21.8%) of 1,541 users in May 2008 (P = 0.005). Of 6,056 pioglitazone users on May 20, 2007, 170 (2.8%) had a CHF diagnosis versus 160 (2.5%) of 6,275 users on December 7, 2007 (P = 0.376), and 122 of 5,998 users in May 2008 (P = 0.006). Although rosiglitazone utilization per million members declined by more than half in 2007, when CV safety concerns started to emerge, about 1 in 5 rosiglitazone users had elevated CV risk at year-end 2007 and in May 2008. About 3% of pioglitazone users in May 2007 had a diagnosis of CHF in claims history, which declined to 2% in May 2008. Insurers should consider the impact of persistent utilization of TZDs among members with CV risk factors when making formulary decisions. |
| 参考文献 |
|
| 其他信息 |
Pioglitazone can cause cancer according to California Labor Code.
Pioglitazone is a member of the class of thiazolidenediones that is 1,3-thiazolidine-2,4-dione substituted by a benzyl group at position 5 which in turn is substituted by a 2-(5-ethylpyridin-2-yl)ethoxy group at position 4 of the phenyl ring. It exhibits hypoglycemic activity. It has a role as an insulin-sensitizing drug, an EC 2.7.1.33 (pantothenate kinase) inhibitor, a xenobiotic, an EC 6.2.1.3 (long-chain-fatty-acid--CoA ligase) inhibitor, a ferroptosis inhibitor, a cardioprotective agent, a PPARgamma agonist, an antidepressant, Pioglitazone hydrochloride is an aromatic ether. Pioglitazone Hydrochloride is the hydrochloride salt of an orally-active thiazolidinedione with antidiabetic properties and potential antineoplastic activity. Pioglitazone activates peroxisome proliferator-activated receptor gamma (PPAR-gamma), a ligand-activated transcription factor, thereby inducing cell differentiation and inhibiting cell growth and angiogenesis. This agent also modulates the transcription of insulin-responsive genes, inhibits macrophage and monocyte activation, and stimulates adipocyte differentiation. A thiazolidinedione and PPAR GAMMA agonist that is used in the treatment of TYPE 2 DIABETES MELLITUS. Pioglitazone is an antihyperglycemic used as an adjunct to diet, exercise, and other antidiabetic medications to manage type 2 diabetes mellitus. It is administered as a racemic mixture, though there is no pharmacologic difference between the enantiomers and they appear to interconvert in vivo with little consequence. The thiazolidinedione class of medications, which also includes [rosiglitazone] and [troglitazone], exerts its pharmacological effect primarily by promoting insulin sensitivity and the improved uptake of blood glucose via agonism at the peroxisome proliferator-activated receptor-gamma (PPARγ). PPARs are ligand-activated transcription factors that are involved in the expression of more than 100 genes and affect numerous metabolic processes, most notably lipid and glucose homeostasis. Thiazolidinediones, including pioglitazone, have fallen out of favor in recent years due to the presence of multiple adverse effects and warnings regarding their use (e.g. congestive heart failure, bladder cancer) and the availability of safer and more effective alternatives for patients with type 2 diabetes mellitus. Pioglitazone is a Peroxisome Proliferator Receptor alpha Agonist, and Peroxisome Proliferator Receptor gamma Agonist, and Thiazolidinedione. The mechanism of action of pioglitazone is as a Peroxisome Proliferator-activated Receptor alpha Agonist, and Peroxisome Proliferator-activated Receptor gamma Agonist. Pioglitazone is an insulin sensitizing agent and thiazolidinedione that is indicated for the treatment of type 2 diabetes. Pioglitazone has been linked to rare instances of acute liver injury. Pioglitazone is an orally-active thiazolidinedione with antidiabetic properties and potential antineoplastic activity. Pioglitazone activates peroxisome proliferator-activated receptor gamma (PPAR-gamma), a ligand-activated transcription factor, thereby inducing cell differentiation and inhibiting cell growth and angiogenesis. This agent also modulates the transcription of insulin-responsive genes, inhibits macrophage and monocyte activation, and stimulates adipocyte differentiation. Pioglitazone is used for the treatment of diabetes mellitus type 2. Pioglitazone selectively stimulates nuclear receptor peroxisone proliferator-activated receptor gamma (PPAR-gamma). It modulates the transcription of the insulin-sensitive genes involved in the control of glucose and lipid metabolism in the lipidic, muscular tissues and in the liver. A thiazolidinedione and PPAR GAMMA agonist that is used in the treatment of TYPE 2 DIABETES MELLITUS. View More
Drug Indication
Therapeutic Uses Hypoglycemic Agents /CLINICAL TRIALS/ ClinicalTrials.gov is a registry and results database of publicly and privately supported clinical studies of human participants conducted around the world. The Web site is maintained by the National Library of Medicine (NLM) and the National Institutes of Health (NIH). Each ClinicalTrials.gov record presents summary information about a study protocol and includes the following: Disease or condition; Intervention (for example, the medical product, behavior, or procedure being studied); Title, description, and design of the study; Requirements for participation (eligibility criteria); Locations where the study is being conducted; Contact information for the study locations; and Links to relevant information on other health Web sites, such as NLM's MedlinePlus for patient health information and PubMed for citations and abstracts for scholarly articles in the field of medicine. Pioglitazone is included in the database. Pioglitazone is used alone (monotherapy) or in combination with a sulfonylurea antidiabetic agent, metformin (either as a fixed-combination preparation or as individual drugs given concurrently), or insulin as an adjunct to diet and exercise for the management of type 2 diabetes mellitus. Pioglitazone is used also in fixed combination with glimepiride in patients with type 2 diabetes mellitus who are already receiving pioglitazone and a sulfonylurea separately or who are inadequately controlled on a sulfonylurea or pioglitazone alone. In patients whose hyperglycemia cannot be controlled with these other antidiabetic agents, pioglitazone should be added to, not substituted for, such antidiabetic therapy. /Included in US product labeling/ /EXPL THER/ Peroxisome proliferator activated receptor gamma-activating drugs show various salutary effects in preclinical models of neurodegenerative disease. The decade-long clinical usage of these drugs as antidiabetics now allows for evaluation of patient-oriented data sources. Using observational data from 2004-2010, we analyzed the association of pioglitazone and incidence of dementia in a prospective cohort study of 145,928 subjects aged >/= 60 years who, at baseline, were free of dementia and insulin-dependent diabetes mellitus. We distinguished between nondiabetics, diabetics without pioglitazone, diabetics with prescriptions of <8 calendar quarters of pioglitazone, and diabetics with =8 quarters. Cox proportional hazard models explored the relative risk (RR) of dementia incidence dependent on pioglitazone use adjusted for sex, age, use of rosiglitazone or metformin, and cardiovascular comorbidities. Long-term use of pioglitazone was associated with a lower dementia incidence. Relative to nondiabetics, the cumulative long-term use of pioglitazone reduced the dementia risk by 47% (RR=0.53, p=0.029). If diabetes patients used pioglitazone <8 quarters, the dementia risk was comparable to those of nondiabetics (RR=1.16, p=0.317), and diabetes patients without a pioglitazone treatment had a 23% increase in dementia risk (RR=1.23, p<0.001). We did not find evidence for age effects, nor for selection into pioglitazone treatment due to obesity. These findings indicate that pioglitazone treatment is associated with a reduced dementia risk in initially non-insulin-dependent diabetes mellitus patients. Prospective clinical trials are needed to evaluate a possible neuroprotective effect in these patients in an ageing population. Drug Warnings /BOXED WARNING/ WARNING: CONGESTIVE HEART FAILURE. Thiazolidinediones, including pioglitazone hydrochloride, cause or exacerbate congestive heart failure in some patients. After initiation of pioglitazone tablets, and after dose increases, monitor patients carefully for signs and symptoms of heart failure (e.g., excessive, rapid weight gain, dyspnea, and/or edema). If heart failure develops, it should be managed according to current standards of care and discontinuation or dose reduction of pioglitazone hydrochloride must be considered. Pioglitazone tablets are not recommended in patients with symptomatic heart failure. Initiation of pioglitazone hydrochloride in patients with established NYHA Class III or IV heart failure is contraindicated. Thiazolidinediones, including pioglitazone, alone or in combination with other antidiabetic agents, can cause fluid retention, which may lead to or exacerbate congestive heart failure (CHF). Use of thiazolidinediones is associated with an approximately twofold increased risk of CHF. Use of pioglitazone in combination with insulin or in patients with New York Heart Association (NYHA) class I or II heart failure may increase the risk. Patients should be observed for signs and symptoms of CHF (e.g., dyspnea, rapid weight gain, edema, unexplained cough or fatigue), especially during initiation of therapy and dosage titration. If signs and symptoms of CHF develop, the disorder should be managed according to current standards of care. In addition, a decrease in the dosage or discontinuance of pioglitazone must be considered in such patients. Patients with New York Heart Association (NYHA) class III or IV cardiac status with or without congestive heart failure (CHF) or with an acute coronary event were not studied in clinical trials of pioglitazone; initiation of therapy with the drug is contraindicated in patients with NYHA class III or IV heart failure. Use of pioglitazone is not recommended in patients with symptomatic heart failure. Caution should be exercised in patients with edema and in those who are at risk for CHF. Thiazolidinedione therapy should not be initiated in hospitalized patients with diabetes mellitus because of the delayed onset of action and because possible drug-related increases in vascular volume and CHF may complicate care of patients with hemodynamic changes induced by coexisting conditions or in-hospital interventions. Risk for pregnancy unless contraceptive measures initiated; anovulatory premenopausal women with insulin resistance may resume ovulation during therapy. The frequency of resumption of ovulation with pioglitazone therapy has not been evaluated in clinical studies, and, therefore, is unknown. If menstrual dysfunction occurs, weigh risks versus benefits of continued pioglitazone. Pharmacodynamics Pioglitazone enhances cellular responsiveness to insulin, increases insulin-dependent glucose disposal, and improves impaired glucose homeostasis. In patients with type 2 diabetes mellitus, these effects result in lower plasma glucose concentrations, lower plasma insulin concentrations, and lower HbA1c values. Significant fluid retention leading to the development/exacerbation of congestive heart failure has been reported with pioglitazone - avoid its use in patients in heart failure or at risk of developing heart failure. There is some evidence that pioglitazone may be associated with an increased risk of developing bladder cancer. Pioglitazone should not be used in patients with active bladder cancer and should be used with caution in patients with a history of bladder cancer. Mechanism of Action Pioglitazone is a selective agonist at peroxisome proliferator-activated receptor-gamma (PPARγ) in target tissues for insulin action such as adipose tissue, skeletal muscle, and liver. Activation of PPARγ increases the transcription of insulin-responsive genes involved in the control of glucose and lipid production, transport, and utilization. Through this mechanism, pioglitazone both enhances tissue sensitivity to insulin and reduces the hepatic production of glucose (i.e. gluconeogenesis) - insulin resistance associated with type 2 diabetes mellitus is therefore improved without an increase in insulin secretion by pancreatic beta cells. DrugBank Repeated administration of peroxisome proliferator-activated receptor gamma (PPARgamma) agonists reduces neuropathic pain-like behavior and associated changes in glial activation in the spinal cord dorsal horn. As PPARgamma is a nuclear receptor, sustained changes in gene expression are widely believed to be the mechanism of pain reduction. However, we recently reported that a single intrathecal (i.t.) injection of pioglitazone, a PPARgamma agonist, reduced hyperalgesia within 30 minutes, a time frame that is typically less than that required for genomic mechanisms. To determine the very rapid antihyperalgesic actions of PPARgamma activation, we administered pioglitazone to rats with spared nerve injury and evaluated hyperalgesia. Pioglitazone inhibited hyperalgesia within 5 minutes of injection, consistent with a nongenomic mechanism. Systemic or i.t. administration of GW9662, a PPARgamma antagonist, inhibited the antihyperalgesic actions of intraperitoneal or i.t. pioglitazone, suggesting a spinal PPAR?-dependent mechanism. To further address the contribution of nongenomic mechanisms, we blocked new protein synthesis in the spinal cord with anisomycin. When coadministered intrathecally, anisomycin did not change pioglitazone antihyperalgesia at an early 7.5-minute time point, further supporting a rapid nongenomic mechanism. At later time points, anisomycin reduced pioglitazone antihyperalgesia, suggesting delayed recruitment of genomic mechanisms. Pioglitazone reduction of spared nerve injury-induced increases in GFAP expression occurred more rapidly than expected, within 60 minutes. We are the first to show that activation of spinal PPARgamma rapidly reduces neuropathic pain independent of canonical genomic activity. We conclude that acute pioglitazone inhibits neuropathic pain in part by reducing astrocyte activation and through both genomic and nongenomic PPARgamma mechanisms. PMID:25599238 Full text: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4329091 Pioglitazone hydrochloride is a thiazolidinedione that depends on the presence of insulin for its mechanism of action. Pioglitazone hydrochloride decreases insulin resistance in the periphery and in the liver resulting in increased insulin-dependent glucose disposal and decreased hepatic glucose output. Pioglitazone is not an insulin secretagogue. Pioglitazone is an agonist for peroxisome proliferator-activated receptor-gamma (PPARgamma). PPAR receptors are found in tissues important for insulin action such as adipose tissue, skeletal muscle, and liver. Activation of PPARgamma nuclear receptors modulates the transcription of a number of insulin responsive genes involved in the control of glucose and lipid metabolism. ... Thiazolidinediones reduce insulin resistance not only in type 2 diabetes but also in non-diabetic conditions associated with insulin resistance such as obesity. The mechanism of action involves binding to the peroxisome proliferator-activated receptor (PPAR)gamma, a transcription factor that regulates the expression of specific genes especially in fat cells but also in other tissues. It is likely that thiazolidinediones primarily act in adipose tissue where PPARgamma is predominantly expressed. Thiazolidinediones have been shown to interfere with expression and release of mediators of insulin resistance originating in adipose tissue (e.g. free fatty acids, adipocytokines such as tumor necrosis factor alpha, resistin, adiponectin) in a way that results in net improvement of insulin sensitivity (i.e. in muscle and liver). Nevertheless, a direct molecular effect in skeletal muscle cannot be excluded. ... PMID:12173692 Pioglitazone, a full peroxisome proliferator-activated receptor (PPAR)-gamma agonist, improves insulin sensitivity by increasing circulating adiponectin levels. However, the molecular mechanisms by which pioglitazone induces insulin sensitization are not fully understood. In this study, we investigated whether pioglitazone improves insulin resistance via upregulation of either 2 distinct receptors for adiponectin (AdipoR1 or AdipoR2) expression in 3T3-L1 adipocytes. Glucose uptake was evaluated by 2-[(3)H] deoxy-glucose uptake assay in 3T3-L1 adipocytes with pioglitazone treatment. AdipoR1 and AdipoR2 mRNA expressions were analyzed by qRT-PCR. /The investigators/ first confirmed that pioglitazone significantly increased insulin-induced 2-deoxyglucose (2-DOG) uptake in 3T3-L1 adipocytes. Next, we investigated the mRNA expression and regulation of AdipoR1 and AdipoR2 after treatment with pioglitazone. Interestingly, pioglitazone significantly induced AdipoR2 expression but it did not affect AdipoR1 expression. In addition, adenovirus-mediated PPARgamma expression significantly enhanced the effects of pioglitazone on insulin-stimulated 2-DOG uptake and AdipoR2 expression in 3T3-L1 adipocytes. These data suggest that pioglitazone enhances adiponectin's autocrine and paracrine actions in 3T3-L1 adipocytes via upregulation of PPARgamma-mediated AdipoR2 expression. Furthermore, we found that pioglitazone significantly increased AMP-activated protein kinase (AMPK) phosphorylation in insulin-stimulated 3T3-L1 adipocytes, but it did not lead to the phosphorylation of IRS-1, Akt, or protein kinase ... Pioglitazone increases insulin sensitivity, at least partly, by PPARgamma-AdipoR2-mediated AMPK phosphorylation in 3T3-L1 adipocytes. In conclusion, the upregulation of AdipoR2 expression may be one of the mechanisms by which pioglitazone improves insulin resistance in 3T3-L1 adipocytes. |
| 分子式 |
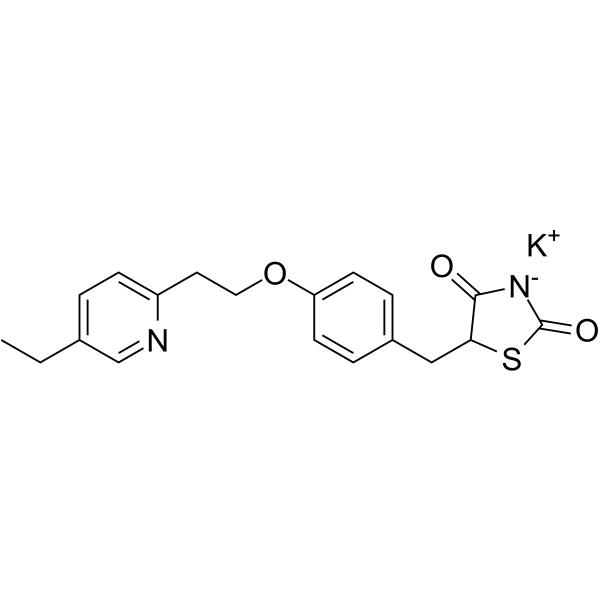
C19H21KN2O3S
|
|---|---|
| 分子量 |
396.544944524765
|
| 精确质量 |
394.075
|
| CAS号 |
1266523-09-4
|
| 相关CAS号 |
Pioglitazone;111025-46-8
|
| PubChem CID |
56653040
|
| 外观&性状 |
Typically exists as solid at room temperature
|
| tPSA |
82.6
|
| 氢键供体(HBD)数目 |
0
|
| 氢键受体(HBA)数目 |
6
|
| 可旋转键数目(RBC) |
7
|
| 重原子数目 |
26
|
| 分子复杂度/Complexity |
472
|
| 定义原子立体中心数目 |
0
|
| SMILES |
C(C1C=CC(OCCC2N=CC(CC)=CC=2)=CC=1)C1SC(=O)NC1=O.[KH]
|
| InChi Key |
YRUUYXLNBAJFIM-UHFFFAOYSA-M
|
| InChi Code |
InChI=1S/C19H20N2O3S.K/c1-2-13-3-6-15(20-12-13)9-10-24-16-7-4-14(5-8-16)11-17-18(22)21-19(23)25-17;/h3-8,12,17H,2,9-11H2,1H3,(H,21,22,23);/q;+1/p-1
|
| 化学名 |
potassium;5-[[4-[2-(5-ethylpyridin-2-yl)ethoxy]phenyl]methyl]-1,3-thiazolidin-3-ide-2,4-dione
|
| 别名 |
Pioglitazone (potassium salt); 1266523-09-4; Pioglitazone potassium; X1ZX7RX9WU; Pioglitazone (potassium); UNII-X1ZX7RX9WU; potassium;5-[[4-[2-(5-ethylpyridin-2-yl)ethoxy]phenyl]methyl]-1,3-thiazolidin-3-ide-2,4-dione; 2,4-Thiazolidinedione, 5-((4-(2-(5-ethyl-2-pyridinyl)ethoxy)phenyl)methyl)-, potassium salt (1:1);
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples
|
|---|---|
| 溶解度 (体内实验) |
注意: 如下所列的是一些常用的体内动物实验溶解配方,主要用于溶解难溶或不溶于水的产品(水溶度<1 mg/mL)。 建议您先取少量样品进行尝试,如该配方可行,再根据实验需求增加样品量。
注射用配方
注射用配方1: DMSO : Tween 80: Saline = 10 : 5 : 85 (如: 100 μL DMSO → 50 μL Tween 80 → 850 μL Saline)(IP/IV/IM/SC等) *生理盐水/Saline的制备:将0.9g氯化钠/NaCl溶解在100 mL ddH ₂ O中,得到澄清溶液。 注射用配方 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (如: 100 μL DMSO → 400 μL PEG300 → 50 μL Tween 80 → 450 μL Saline) 注射用配方 3: DMSO : Corn oil = 10 : 90 (如: 100 μL DMSO → 900 μL Corn oil) 示例: 以注射用配方 3 (DMSO : Corn oil = 10 : 90) 为例说明, 如果要配制 1 mL 2.5 mg/mL的工作液, 您可以取 100 μL 25 mg/mL 澄清的 DMSO 储备液,加到 900 μL Corn oil/玉米油中, 混合均匀。 View More
注射用配方 4: DMSO : 20% SBE-β-CD in Saline = 10 : 90 [如:100 μL DMSO → 900 μL (20% SBE-β-CD in Saline)] 口服配方
口服配方 1: 悬浮于0.5% CMC Na (羧甲基纤维素钠) 口服配方 2: 悬浮于0.5% Carboxymethyl cellulose (羧甲基纤维素) 示例: 以口服配方 1 (悬浮于 0.5% CMC Na)为例说明, 如果要配制 100 mL 2.5 mg/mL 的工作液, 您可以先取0.5g CMC Na并将其溶解于100mL ddH2O中,得到0.5%CMC-Na澄清溶液;然后将250 mg待测化合物加到100 mL前述 0.5%CMC Na溶液中,得到悬浮液。 View More
口服配方 3: 溶解于 PEG400 (聚乙二醇400) 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.5218 mL | 12.6088 mL | 25.2175 mL | |
| 5 mM | 0.5044 mL | 2.5218 mL | 5.0435 mL | |
| 10 mM | 0.2522 mL | 1.2609 mL | 2.5218 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
| NCT Number | Recruitment | interventions | Conditions | Sponsor/Collaborators | Start Date | Phases |
| NCT04501406 | Recruiting | Drug: Pioglitazone Other: Placebo |
Type 2 Diabetes Mellitus (T2DM) Nonalcoholic Steatohepatitis |
University of Florida | December 15, 2020 | Phase 2 |
| NCT01873001 | Completed | Drug: Pioglitazone HCl Drug: Abiraterone acetate |
Healthy Volunteers | Janssen Research & Development, LLC | May 2013 | Phase 1 |
| NCT02958956 | Completed Has Results | Drug: Pioglitazone | Diabetes Mellitus, Type 2, Cancer | Takeda | January 1, 1997 | |
| NCT03080480 | Terminated | Drug: Pioglitazone | Chronic Granulomatous Disease | Children's Hospital of Fudan University | September 1, 2017 | Phase 1 Phase 2 |
 AOH-1996
AOH-1996
 hMAO-B-IN-3
hMAO-B-IN-3
 Flupyrimin
Flupyrimin
 阿曲库铵
阿曲库铵
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1





























