
| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 100mg |
|
||
| 500mg |
|
||
| 1g |
|
||
| 2g |
|
||
| 5g |
|
||
| 10g |
|
||
| 100g |
|
||
| Other Sizes |
|

| 靶点 |
Voltage-gated Na+ channels (VGSCs)
|
|---|---|
| 体外研究 (In Vitro) |
一种抗氧化药物是苯妥英。对于缺席计划或肌肉基质补剂等原有的综合优势无效,但对于局部和综合效果都强大的直接基质补剂有帮助。据信苯妥英通过电压门控实现这一点。可以通过电压阻塞通道来停止编程 [2]。苯妥英表现出对大腿上膜电位的静息通道的低神经元亲和力[3]。当通道和中期上部变为开放、非活动状态时,会发生更多的结合和阻塞。由于阻塞效应非常依赖于使用情况,因此它会在长期或频繁激活后(例如发生引用时)建立起来。由于苯妥英对钠通道的阻断作用缓慢,因此它不会快速改变当前的时间进程或爆发由普通持续时间的突触分解的最强事件电位。因此,在不显着影响发作活动的情况下,苯妥英偶尔可以降低阶段患者的病理性过度兴奋。此外,苯妥英会导致电流持续爆发,这对于管理工程数据可能至关重要。一种 1b 类抗心律失常药物是苯妥英[4]。
|
| 体内研究 (In Vivo) |
苯妥英(5,5-二苯基乙内酰脲;60 mg/kg;每天;28 天)通过使用 MDA-MB-231 细胞减少六周龄雌性 Rag2-/-Il2rg-/- 小鼠肿瘤的形成 [1] 。
边缘癫痫发生改变了苯妥英在Sprague-Dawley大鼠中的抗惊厥作用[6]。 |
| 酶活实验 |
苯妥英被发现与钠通道的快速失活状态紧密结合,但结合发生缓慢,这是苯妥英破坏癫痫放电而对正常放电活动影响最小的一个关键特征。[5]
抗惊厥苯妥英抑制大鼠海马神经元的Na+电流,其效力在去极化保持电位下显著增加,表明与静息Na+通道的结合较弱,但与开放或失活的通道的结合较紧密。四种不同的实验测量,即在不同保持电位下的稳定阻滞,在去极化保持电位下的打开和关闭动力学,失活曲线的移动,以及从失活中恢复的剂量依赖性减慢,估计苯妥英与失活通道结合的Kd约为7微米。苯妥英治疗浓度的显著阻滞需要至少几秒钟的长时间去极化。阻滞的缓慢发展并不反映苯妥英与通道缓慢失活状态的选择性结合,因为阻滞的发展速度比缓慢失活更快,需要的去极化电压更少。相反,苯妥英似乎紧密但缓慢地结合(大约10(4)M-1秒-1)到Na+通道的快速失活状态。这种紧密但缓慢的结合可能是苯妥英破坏癫痫放电而对正常放电模式影响最小的能力的基础。[5] |
| 细胞实验 |
本研究考察了苯妥英钠对小鼠胸锁乳突神经肌肉组织中e.p.ps量含量的影响。暴露于含有苯妥英钠(10 pg/ml)的溶液时,e.p.ps的平均振幅降低。结果表明,苯妥英钠浓度显著降低了m.e.p.cs的衰减时间常数,但对衰减幅度影响不大。在苯妥英的存在下,m.e.p.cs的衰变似乎缩短了m.e.p.cs的生长时间。在三个实验中,生长时间从对照溶液中的175 + 19 ms下降到苯妥英溶液中的146 + 10 ms。A.结果表明苯妥英在神经肌肉交界处有两种类型的抑制作用。[7]
|
| 动物实验 |
We have previously reported that the VGSC-blocking antiepileptic drug phenytoin inhibits the migration and invasion of metastatic MDA-MB-231 cells in vitro. The purpose of the present study was to establish whether VGSCs might be viable therapeutic targets by testing the effect of phenytoin on tumour growth and metastasis in vivo. We found that expression of Nav1.5, previously detected in MDA-MB-231 cells in vitro, was retained on cells in orthotopic xenografts. Treatment with phenytoin, at a dose equivalent to that used to treat epilepsy (60 mg/kg; daily), significantly reduced tumour growth, without affecting animal weight. Phenytoin also reduced cancer cell proliferation in vivo and invasion into surrounding mammary tissue. Finally, phenytoin significantly reduced metastasis to the liver, lungs and spleen.
Conclusions: This is the first study showing that phenytoin reduces breast tumour growth and metastasis in vivo. We propose that pharmacologically targeting VGSCs, by repurposing antiepileptic or antiarrhythmic drugs, should be further studied as a potentially novel anti-cancer therapy.[1]
Studies on the anticonvulsant efficacy of the major antiepileptic drug phenytoin in kindled rats have often reported inconsistent effects. It has been proposed that technical and genetic factors or poor and variable absorption of phenytoin after i.p. or oral administration may be involved in the lack of consistent anticonvulsant activity of phenytoin in this model of temporal lobe epilepsy. We examined if kindling itself changes the anticonvulsant efficacy of phenytoin by testing this drug before and after amygdala kindling in male and female Sprague-Dawley rats. To exclude the possible bias of poor and variable absorption, blood was sampled in all experiments for drug analysis in plasma. The threshold for induction of focal seizures (afterdischarge threshold; ADT) was used for determining phenytoin's anticonvulsant activity. Before kindling, phenytoin, 75 mg/kg i.p., markedly increased ADT in both genders, although the effect was more pronounced in males. Following kindling, the anticonvulsant activity obtained with phenytoin, 75 mg/kg, before kindling was totally lost, and female rats even exhibited a proconvulsant effect upon administration of this dose, indicating that kindling had dramatically altered the anticonvulsant efficacy of phenytoin. Plasma levels of phenytoin were comparable before and after kindling, and were within or near to the 'therapeutic range' known from epileptic patients. When the dose of phenytoin was reduced to 50 or 25 mg/kg i.p., significant anticonvulsant effects on ADT were obtained. When phenytoin, 50 mg/kg, was administered i.p. or i.v. in the same group of fully kindled rats, both anticonvulsant activity and plasma drug levels were comparable with both routes, indicating that the i.p. route is suited for such studies. The data indicate that kindling alters the dose-response of phenytoin in that a high anticonvulsant dose becomes ineffective or proconvulsant after kindling, possibly by an increased sensitivity of the kindled brain to proconvulsant effects of phenytoin which normally only occur at much higher doses. If similar alterations evolve in humans during development of chronic epilepsy, this may be involved in the mechanisms leading to intractability of temporal lobe epilepsy.[6] |
| 药代性质 (ADME/PK) |
Absorption, Distribution and Excretion
Given its narrow therapeutic index, therapeutic drug monitoring is recommended to help guide dosing. Phenytoin is completely absorbed. Peak plasma concentration is attained approximately 1.5-3 hours, and 4-12 hours after administration of the immediate release formulation and the extended release formulation, respectively. It should be noted that absorption can be markedly prolonged in situations of acute ingestion. The majority of phenytoin is excreted as inactive metabolites in the bile. An estimated 1-5% of phenytoin is eliminated unchanged in the urine. The volume of distribution of phenytoin is reported to be approximately 0.75 L/kg. The clearance of phenytoin is non-linear. At lower serum concentrations (less than 10 mg/L), elimination is characterized by first order kinetics. As plasma concentrations increase, the kinetics shift gradually towards zero-order, and finally reach zero-order kinetics once the system is saturated. Studies using Dilantin have shown that phenytoin and its sodium salt are usually completely absorbed from the GI tract. Bioavailability may vary enough among oral phenytoin sodium preparations of different manufacturers to result in toxic serum concentrations or a loss of seizure control (subtherapeutic serum concentrations)... Absorption of phenytoin is slow and variable among products (poor in neonates) for oral admininstration, immediate for iv administration, and very slow but complete (92%) for intramuscular administration. Prompt phenytoin capsules are rapidly absorbed and generally produce peak serum concentrations in 1.5-3 hours, while extended phenytoin sodium capsules are more slowly absorbed and generally produce peak serum concentrations in 4-12 hours. When phenytoin sodium is administered im, absorption may be erratic; this may result from crystallization of the drug at the injection site because of the change in pH. /Phenytoin/ is distributed into cerebrospinal fluid, saliva, semen, GI fluids, bile, and breast milk; it also crosses the placenta, with fetal serum concentrations equal to those of the mother. For more Absorption, Distribution and Excretion (Complete) data for PHENYTOIN (15 total), please visit the HSDB record page. Metabolism / Metabolites Phenytoin is extensively metabolized and is first transformed into a reactive _arene oxide intermediate_. It is thought that this reactive intermediate is responsible for many undesirable phenytoin adverse effects such as hepatotoxicity, SJS/TEN, and other idiosyncratic reactions. The _arene oxide_ is metabolized to either a _hydroxyphenytoin_ or _phenytoin dihydrodiol_ metabolite, although the former accounts for about 90% of phenytoin metabolism. Interestingly, two stereoisomers of the _hydroxyphenytoin_ metabolite are formed by CYP2C9 and CYP2C19: _(R)-p-HPPH_ and _(S)-p-HPPH_. When CYP2C19 catalyzes the reaction, the ratio of stereoisomers is roughly 1:1, whereas when CYP2C9 catalyzes the reaction, the ratio heavily favours the "S" stereoisomer. Since the metabolism of phenytoin is in part influenced by genetic polymorphisms of CYP2C9 and CYP2C19, this ratio can be utilized to identify different genomic variants of the enzymes. EPHX1, CYP1A2, CYP2A6, CYP2C19, CYP2C8, CYP2C9, CYP2D6, CYP2E1 and CYP3A4 are responsible for producing the _phenytoin dihydrodiol_ metabolite. _Hydroxyphenytoin_ can be metabolized by CYP2C19, CYP3A5, CYP2C9, CYP3A4, CYP3A7, CYP2B6 and CYP2D6 to a _phenytoin catechol_ metabolite or undergo glucuronidation by UGT1A6, UGT1A9, UGT1A1, and UGT1A4 to a _glucuronide metabolite_ that can be eliminated in the urine. On the other hand, the _phenytoin dihydrodiol_ entity is only transformed to the _catechol_ metabolite. The _catechol metabolite_ can undergo methylation by COMT and be subsequently eliminated in the urine, or can spontaneously oxidize to a _phenytoin quinone_ (NQO1 can transform the quinone back to the catechol metabolite). Of note, although CYP2C18 is poorly expressed in the liver, the enzyme is active in the skin and is involved in the primary and secondary hydroxylation of phenytoin. This CYP2C18 mediated bioactivation may be linked to the manifestation of adverse cutaneous drug reactions associated with phenytoin. The major route of metabolism of phenytoin is oxidation by the liver to the inactive metabolite 5-(p-hydroxyphenyl)-5-phenylhydantoin (HPPH). Because this metabolism is a saturable process, small increases in dosage may produce substantial increases in plasma phenytoin concentrations... The rate of hepatic biotransformation is increased in younger children, in pregnant women, in women during menses, and in patients with acute trauma; rate decreases with advancing age. The major inactive metabolite of phenytoin is 5-(p-hydroxyphenyl)-5-phenylhydantoin (HPPH). Phenytoin may be metabolized slowly in a small number of individuals due to genetic predisposition, which may cause limited enzyme availability and lack of induction. ... Oxidative metabolism of 1 of geminal phenyl rings of diphenylhydantoin ... 5-meta-hydroxyphenyl-(l) and 5-para-hydroxyphenyl-5-phenylhydantoin were detected in urine of man (approx ratio 1:12) ... Phenytoin has known human metabolites that include 3'-HPPH, 4-Hydroxyphenytoin, 5-(3,4-dihydroxycyclohexa-1,5-dien-1-yl)-5-phenylimidazolidine-2,4-dione, and (2S,3S,4S,5R)-6-(2,5-dioxo-4,4-diphenylimidazolidin-1-yl)-3,4,5-trihydroxyoxane-2-carboxylic acid. Primarily hepatic. The majority of the dose (up to 90%) is metabolized to 5-(4'-hydroxyphenyl)-5-phenylhydantoin (p-HPPH). This metabolite undergoes further glucuronidation and is excreted into the urine. CYP2C19 and CYP2C9 catalyze the aforementioned reaction. Route of Elimination: Most of the drug is excreted in the bile as inactive metabolites which are then reabsorbed from the intestinal tract and excreted in the urine. Urinary excretion of phenytoin and its metabolites occurs partly with glomerular filtration but, more importantly, by tubular secretion. Half Life: 22 hours (range of 7 to 42 hours) Biological Half-Life Oral administration: The half-life of phenytoin ranges from 7 to 42 hours, and is 22 hours on average. Intravenous administration: The half-life of phenytoin ranges from 10-15 hours. Following oral administration, the plasma half-life of phenytoin averages about 22 hours, although the half-life has ranged from 7-42 hours in individual patients. The plasma half-life of phenytoin in humans following IV administration ranges from 10-15 hours. Because phenytoin exhibits saturable, zero-order, or dose-dependent pharmacokinetics, the apparent half-life of phenytoin changes with dose and serum concentrations. this is due to the saturation of the enzyme system responsible for metabolizing phenytoin, which occurs at therapeutic concentrations of the drug. Thus, a constant amount of drug is metabolized (capacity-limited metabolism), and small increases in dose may cause disproportionately large increases in serum concentrations and apparent half-life, possibly causing unexpected toxicity. |
| 毒性/毒理 (Toxicokinetics/TK) |
Hepatotoxicity
Prospective studies indicate that a fairly high proportion of patients taking phenytoin have transient serum aminotransferase elevations. These elevations are usually benign, not associated with liver histological abnormalities and usually resolve even with drug continuation. In addition, a higher proportion of patients have mild-to-moderate elevations in gammaglutamyl transpeptidase (GGT) levels, which is indicative of hepatic enzyme induction rather than liver injury. Marked aminotransferase elevations (>3 fold elevated) occur rarely. Importantly, however, phenytoin is one of the most common causes of clinically apparent drug induced liver disease and acute liver failure. More than 100 cases of liver injury due to phenytoin (diphenylhydantoin) have been published and a characteristic clinical pattern (signature) of injury has been described. The estimated frequency ranges from 1 per 1000 to 1 per 10,000 and probably varies by race and ethnicity. The typical case arises after 2 to 8 weeks of therapy with initial onset of fever, rash, facial edema and lymphadenopathy, followed in a few days by jaundice and dark urine. The serum enzyme elevations can be hepatocellular, although mixed patterns are probably more common and rare cases are cholestatic. Eosinophilia, increased white counts and atypical lymphocytosis are also common. Autoantibody formation is rare. The clinical symptoms and signs can mimic acute mononucleosis or even lymphoma (pseudo-lymphoma syndrome). Almost all cases of phenytoin hepatotoxicity occur in the context of a systemic hypersensitivity syndrome and it is referred to often as the anticonvulsant hypersensitivity syndrome (HDS) or drug rash with eosinophilia and systemic symptoms syndrome (DRESS). Other manifestations can be Stevens-Johnson syndrome, toxic epidermal necrolysis, aplastic anemia, thrombocytopenia, neutropenia, nephritis, and pneumonitis. Most cases of liver injury are self-limiting and resolve within 1 to 2 months of stopping phenytoin. However, the liver injury can be severe and many fatal instances have been reported, phenytoin usually appearing in the top 10 causes of drug induced acute liver failure. In the typical case, however, recovery is usually complete. Likelihood score: A (well known cause of clinically apparent liver injury). Effects During Pregnancy and Lactation ◉ Summary of Use during Lactation Breastfeeding during phenytoin monotherapy does not appear to adversely affect infant growth or development, and breastfed infants had higher IQs and enhanced verbal abilities than nonbreastfed infants at 6 years of age in one study. If phenytoin is required by the mother, it is not a reason to discontinue breastfeeding. Because of the low levels of phenytoin in breastmilk, amounts ingested by the infant are small and usually cause no difficulties in breastfed infants when used alone except for rare idiosyncratic reactions. Combination therapy with sedating anticonvulsants or psychotropics may result in infant sedation or withdrawal reactions. In one case report, maternal phenytoin dosage requirements decreased as breastfeeding was discontinued. ◉ Effects in Breastfed Infants A mother was taking phenobarbital 390 mg daily and phenytoin 400 mg daily during pregnancy and postpartum. Her infant was drowsy at birth, refused to suck and was given partial formula feeding. At 5 days of age, her infant was admitted to the hospital pale and collapsed with bruising, bleeding, and a decreased hemoglobin, thought to be due to methemoglobinemia. Breastfeeding was discontinued and the infant was given a transfusion which rapidly improved her condition. On day 10, the mother resumed breastfeeding the infant. Within 24 hours the infant was extremely sedated and refused to suck and was fed breastmilk with a spoon. The sedation persisted for 2 days until breastmilk was discontinued permanently because of a return of methemoglobinemia. The extreme sedation was probably due to phenobarbital in the milk and the methemoglobinemia was probably caused by the phenytoin. One clinician reported that the breastfed infants of 28 mothers who were taking phenytoin 100 to 200 mg 3 times daily had no adverse reactions including drowsiness or lethargy. No adverse effects were noted in the breastfed neonates of 2 mothers who were taking phenytoin 300 mg daily. A 10-week-old breastfed infant whose mother was taking clemastine, phenytoin and carbamazepine was drowsy, refused to feed, was irritable, and had high-pitched crying. These side effects were possibly caused by clemastine in breastmilk, but the other drugs could also have contributed. A probable case of drug-induced drowsiness occurred in a newborn whose mother was taking primidone, carbamazepine and phenytoin (dosages not stated). On day 30, breastfeeding was discontinued because of the drowsiness that occurred after each feeding and poor weight gain. The same group of researchers found that 15 partially breastfed infants whose mothers were taking various anticonvulsants, including phenytoin, gained weight at a slower rate during the first 5 days postpartum than did 75 infants of epileptic mothers who bottle fed or control mothers taking no medications. Drowsiness, pallor and feeding difficulties in a 2-week-old were possibly caused by primidone and phenytoin in breastmilk. Possible drug-related drowsiness, pallor and feeding difficulties were reported in a 4-day-old whose mother was taking primidone, phenobarbital, phenytoin and sulthiame. Although phenytoin might have contributed to these outcomes, it is more likely that they were due primarily to the more sedating anticonvulsants, primidone and phenobarbital. Two breastfed infants (one full, one partial) whose mothers took phenytoin during pregnancy and postpartum became hyperexcitable when their serum phenytoin dropped to unmeasurable levels at 3 to 6 weeks of age. In a long-term study on infants exposed to anticonvulsants during breastfeeding, no difference in average intelligence quotient at 3 years of age was found between infants who were breastfed (n = 17) a median of 6 months and those not breastfed (n = 23) when their mothers were taking phenytoin. At 6 years of age, extensive psychological and intelligence testing found no difference between the breastfed and nonbreastfed infants. ◉ Effects on Lactation and Breastmilk Relevant published information was not found as of the revision date. Protein Binding Phenytoin is roughly 90% protein bound. |
| 参考文献 |
[1] The sodium channel-blocking antiepileptic drug phenytoin inhibits breast tumour growth and metastasis. Mol Cancer. 2015 Jan 27;14(1):13.
[2]. The neurobiology of antiepileptic drugs. Nat Rev Neurosci, 2004. 5(7): p. 553-64. [3]. Mechanisms of action of antiseizure drugs. Handb Clin Neurol, 2012. 108: p. 663-81. [4]. Medical therapy for sudden death. Pediatr Clin North Am, 2004. 51(5): p. 1379-87. [5]. Slow binding of phenytoin to inactivated sodium channels in rat hippocampal neurons. Mol. Pharmacol. 46, 716–725 (1994). [6]. Limbic epileptogenesis alters the anticonvulsant efficacy of phenytoin in Sprague-Dawley rats. Epilepsy Res. 31, 175–186 (1998). [7]. Presynaptic and postsynaptic depressant effects of phenytoin sodium at the neuromuscular junction. Br J Pharmacol . 1980 May;69(1):119-21. |
| 其他信息 |
Diphenylhydantoin (Phenytoin) can cause cancer and developmental toxicity according to an independent committee of scientific and health experts.
Phenytoin appears as fine white or almost white crystalline powder. Odorless or almost odorless. Tasteless. (NTP, 1992) Phenytoin is a imidazolidine-2,4-dione that consists of hydantoin bearing two phenyl substituents at position 5. It has a role as an anticonvulsant, a teratogenic agent, a drug allergen and a sodium channel blocker. It is functionally related to a hydantoin. Phenytoin is classified as a hydantoin derivative and despite its narrow therapeutic index, it is one of the most commonly used anticonvulsants. Since it's introduction about 80 years ago, phenytoin has not only been established as an effective anti-epileptic, but has also been investigated for several other indications such as bipolar disorder, retina protection, and wound healing. Clinicians are advised to initiate therapeutic drug monitoring in patients who require phenytoin since even small deviations from the recommended therapeutic range can lead to suboptimal treatment, or adverse effects. Both parenteral and oral formulations of phenytoin are available on the market. Phenytoin is an Anti-epileptic Agent. The mechanism of action of phenytoin is as a Cytochrome P450 1A2 Inducer, and Cytochrome P450 2B6 Inducer, and Cytochrome P450 2C8 Inducer, and Cytochrome P450 2C19 Inducer, and Cytochrome P450 2D6 Inducer, and Cytochrome P450 3A Inducer, and Cytochrome P450 2C9 Inducer. The physiologic effect of phenytoin is by means of Decreased Central Nervous System Disorganized Electrical Activity. Phenytoin, formerly known as diphenylhydantoin, is a potent anticonvulsant used to treat and prevent generalized grand mal seizures, complex partial seizures and status epilepticus. Phenytoin was formerly the most commonly used anticonvulsant agent but is now declining in use, having been replaced by more modern, better tolerated agents. Phenytoin is an uncommon but well known cause of acute idiosyncratic drug induced liver disease that can be severe and even fatal. Phenytoin Sodium is the sodium salt form of phenytoin, a hydantoin derivate and non-sedative antiepileptic agent with anticonvulsant activity. Phenytoin sodium promotes sodium efflux from neurons located in the motor cortex, thereby stabilizing the neuron and inhibiting synaptic transmission. This leads to a reduction in posttetanic potentiation at synapses, an inhibition of repetitive firing of action potentials and ultimately inhibits the spread of seizure activity. Phenytoin is a hydantoin derivative and a non-sedative antiepileptic agent with anticonvulsant activity. Phenytoin potentially acts by promoting sodium efflux from neurons located in the motor cortex reducing post-tetanic potentiation at synapses. The reduction of potentiation prevents cortical seizure foci spreading to adjacent areas, stabilizing the threshold against hyperexcitability. In addition, this agent appears to reduce sensitivity of muscle spindles to stretch causing muscle relaxation. An anticonvulsant that is used in a wide variety of seizures. It is also an anti-arrhythmic and a muscle relaxant. The mechanism of therapeutic action is not clear, although several cellular actions have been described including effects on ion channels, active transport, and general membrane stabilization. The mechanism of its muscle relaxant effect appears to involve a reduction in the sensitivity of muscle spindles to stretch. Phenytoin has been proposed for several other therapeutic uses, but its use has been limited by its many adverse effects and interactions with other drugs. An anticonvulsant that is used to treat a wide variety of seizures. It is also an anti-arrhythmic and a muscle relaxant. The mechanism of therapeutic action is not clear, although several cellular actions have been described including effects on ion channels, active transport, and general membrane stabilization. The mechanism of its muscle relaxant effect appears to involve a reduction in the sensitivity of muscle spindles to stretch. Phenytoin has been proposed for several other therapeutic uses, but its use has been limited by its many adverse effects and interactions with other drugs. See also: Phenytoin Sodium (annotation moved to). Drug Indication Phenytoin is indicated to treat grand mal seizures, complex partial seizures, and to prevent and treat seizures during or following neurosurgery. Injectable phenytoin and [Fosphenytoin], which is the phosphate ester prodrug formulation of phenytoin, are indicated to treat tonic-clonic status epilepticus, and for the prevention and treatment of seizures occurring during neurosurgery. Mechanism of Action Although phenytoin first appeared in the literature in 1946, it has taken decades for the mechanism of action to be more specifically elucidated. Although several scientists were convinced that phenytoin altered sodium permeability, it wasn’t until the 1980’s that this phenomenon was linked to voltage-gated sodium channels. Phenytoin is often described as a non-specific sodium channel blocker and targets almost all voltage-gated sodium channel subtypes. More specifically, phenytoin prevents seizures by inhibiting the positive feedback loop that results in neuronal propagation of high frequency action potentials. The mechanism of action is not completely known, but it is thought to involve stabilization of neuronal membranes at the cell body, axon, and synapse and limitation of the spread of neuronal or seizure activity. In neurons, phenytoin decreases sodium and calcium ion influx by prolonging channel inactivation time during generation of nerve impulses. Phenytoin blocks the voltage-dependant sodium channels of neurons and inhibits the calcium flux across neuronal membranes, thus helping to stabilize neurons. It also decreases synaptic transmission, and decreases post-tetanic potentiation at the synapse. Phenytoin enhances the sodium ATPase activity of neurons and/or glial cells. It also influences second messenger systems by inhibiting calcium-calmodulin protein phosphorylation and possibly altering cyclic nucleotide production or metabolism. Phenytoin may act to normalize influx of sodium and calcium to cardiac Purkinje fibers. Abnormal ventricular automaticity and membrane responsiveness are decreased. Also, phenytoin shortens the refractory period, and therefore shortens the QT interval and the duration of the action potential. Exact mechanism is unknown. Phenytoin may act in the CNS to decrease synaptic transmission or to decrease summation of temporal stimulation leading to neuronal discharge (antikindling). Phenytoin raises the threshold of facial pain and shortens the duration of attacks by diminishing self-maintenance of excitation and repetitive firing. Phenytoin's mechanisms of action as a muscle relaxant is thought to be similar to its anticonvulsant action. In movement disorders, the membrane stabilizing effect reduces abnormal sustained repetitive firing and potentiation of nerve and muscle cells. A number of studies suggest that keratinocyte growth factor (KGF) plays a major part in reepithelialization after injury, via binding to the specific KGF receptor (KGFR). Several pharmacological agents, including the anti-epileptic drug phenytoin (PHT), have been widely used clinically to promote wound healing. Although the mechanism of action of PHT in this process is still not well understood, it is possible that the activity of PHT in wound healing is mediated via KGF and the KGFR. In the present study, using the enzyme-linked immunosorbant assay and flow cytometry we have shown that PHT increases KGF secretion and KGFR expression by more than 150% in gingival fibroblasts and epithelial cells, respectively. Moreover, semi-quantitative reverse transcriptase-polymerase chain reaction analysis showed that PHT also markedly increased both KGF and KGFR gene transcription by these cells. |
| 分子式 |
C₁₅H₁₂N₂O₂
|
|
|---|---|---|
| 分子量 |
252.27
|
|
| 精确质量 |
252.089
|
|
| 元素分析 |
C, 71.42; H, 4.79; N, 11.10; O, 12.68
|
|
| CAS号 |
57-41-0
|
|
| 相关CAS号 |
Phenytoin sodium;630-93-3;Phenytoin-d10;65854-97-9;Phenytoin-15N2,13C;78213-26-0; 57-41-0
|
|
| PubChem CID |
1775
|
|
| 外观&性状 |
White to off-white solid powder
|
|
| 密度 |
1.3±0.1 g/cm3
|
|
| 沸点 |
464.0±55.0 °C at 760 mmHg
|
|
| 熔点 |
293-295 °C(lit.)
|
|
| 闪点 |
305.8±20.8 °C
|
|
| 蒸汽压 |
0.0±1.2 mmHg at 25°C
|
|
| 折射率 |
1.652
|
|
| LogP |
2.29
|
|
| tPSA |
58.2
|
|
| 氢键供体(HBD)数目 |
2
|
|
| 氢键受体(HBA)数目 |
2
|
|
| 可旋转键数目(RBC) |
2
|
|
| 重原子数目 |
19
|
|
| 分子复杂度/Complexity |
350
|
|
| 定义原子立体中心数目 |
0
|
|
| InChi Key |
CXOFVDLJLONNDW-UHFFFAOYSA-N
|
|
| InChi Code |
InChI=1S/C15H12N2O2/c18-13-15(17-14(19)16-13,11-7-3-1-4-8-11)12-9-5-2-6-10-12/h1-10H,(H2,16,17,18,19)
|
|
| 化学名 |
2,4-Imidazolidinedione, 5,5-diphenyl-
|
|
| 别名 |
|
|
| HS Tariff Code |
2934.99.9001
|
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
|
|||
|---|---|---|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 2.5 mg/mL (9.91 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 25.0 mg/mL澄清DMSO储备液加入到400 μL PEG300中,混匀;然后向上述溶液中加入50 μL Tween-80,混匀;加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: ≥ 2.5 mg/mL (9.91 mM) (饱和度未知) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 25.0 mg/mL澄清DMSO储备液加入900 μL 20% SBE-β-CD生理盐水溶液中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 View More
配方 3 中的溶解度: ≥ 2.5 mg/mL (9.91 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 3.9640 mL | 19.8200 mL | 39.6401 mL | |
| 5 mM | 0.7928 mL | 3.9640 mL | 7.9280 mL | |
| 10 mM | 0.3964 mL | 1.9820 mL | 3.9640 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
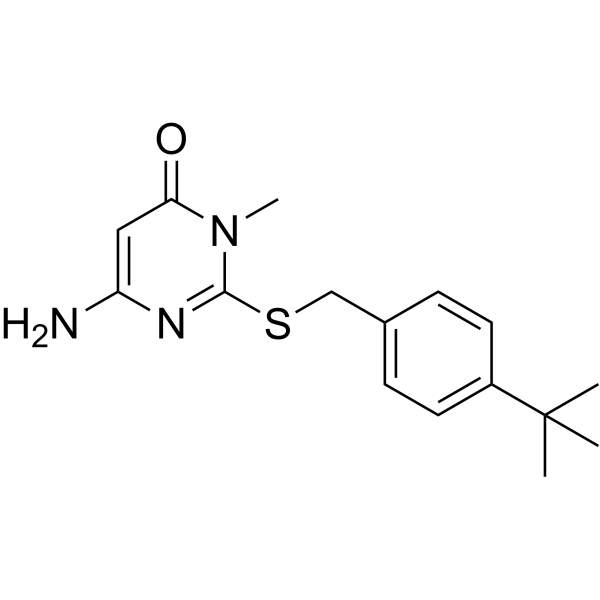 Nav1.7-IN-15
Nav1.7-IN-15
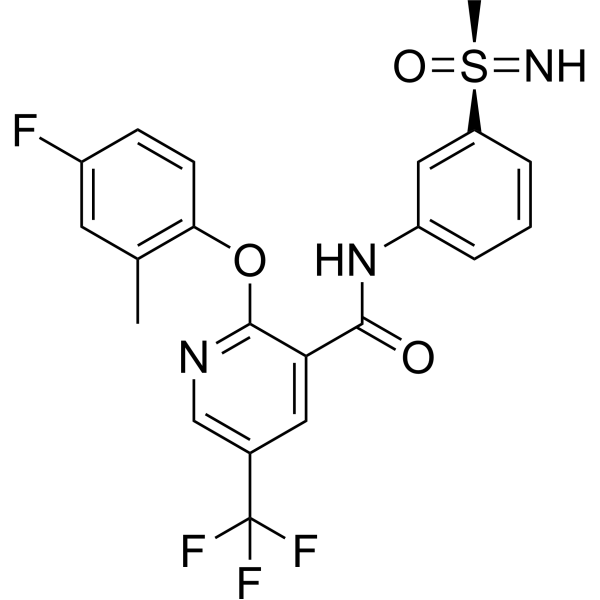 (S)-LTGO-33
(S)-LTGO-33
 Nav1.8-IN-15
Nav1.8-IN-15
 Crotamine TFA
Crotamine TFA
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1





























 COA
COA
