| 规格 | 价格 | |
|---|---|---|
| 500mg | ||
| 1g | ||
| Other Sizes |
| 靶点 |
ALKL1196 (IC50 = 15-43 nM); ALKG1269A (IC50 = 14-80 nM); ALK1151Tins (IC50 = 38-50 nM); ALKG1202R (IC50 = 77-113 nM); ALKWT (IC50 <0.07 nM); ALKL1996M (IC50 = 0.6 nM); ALKG1269A (IC50 = 0.9 nM); ALK1151Tins (IC50 = 0.1 nM); ALKL1152R (IC50 <0.1 nM); ALKS1206Y (IC50 = 0.2 nM); ALKC1156Y (IC50 <0.1 nM); ALKF1174L (IC50 <0.1nM)
|
|---|---|
| 体外研究 (In Vitro) |
体外活性:PF-06463922 表现出针对 ALK 和大量 ALK 临床突变的显着细胞活性,IC50 范围为 0.2 nM-77 nM。 PF-06463922 显着抑制含有 SLC34A2-ROS1 融合的 HCC78 人 NSCLC 细胞和表达人 CD74-ROS1 的 BaF3-CD74-ROS1 细胞的细胞增殖并诱导细胞凋亡。 PF-06463922 还显示出有效的生长抑制活性,并诱导含有非突变 ALK 或突变 ALK 融合的 NSCLC 细胞凋亡。 激酶测定:重组人野生型和突变 ALK 激酶结构域蛋白(氨基酸 1093-1411)在-house使用杆状病毒表达,通过MgATP自磷酸化预激活,并使用微流体迁移率变化测定法测定激酶活性。反应含有 1.3 nM 野生型 ALK 或 0.5 nM 突变型 ALK(适合反应 1 小时后肽底物产生 15-20% 磷酸化)、3 μM 5-FAM-KKSRGDYMTMQIG-CONH2)、5 mM MgCl2 和25 mM Hepes(pH 7.1)中 ATP 的 Km 水平。动力学和晶体学研究显示该抑制剂具有 ATP 竞争性。 Ki 值是通过将转化率 (%) 拟合到竞争性抑制方程来计算的。 ROS1 酶的测定方法如上所述,但使用 0.25 nM 重组人 ROS1 催化结构域(氨基酸 1883-2347)。使用 206 激酶组评估激酶抑制剂的选择性。细胞测定:将细胞接种在含有 10% FBS 的生长培养基中的 96 孔板中,并在 37°C 下培养过夜。第二天,将连续稀释的 Lorlatinib 或适当的对照添加到指定孔中,并将细胞在 37°C 下孵育 72 小时。进行 CellTiter-Glo 测定以确定相对细胞数。 IC50值通过使用四参数分析方法的浓度-响应曲线拟合来计算。
|
| 体内研究 (In Vivo) |
如前所述,LSL-FIG-ROS1;Cdkn2a−/−;LSL-Luc 小鼠通过颅内立体定向注射 Adeno-Cre 启动 GBM 肿瘤的从头发生。如下所述,使用 BLI 监测肿瘤发展。一旦肿瘤达到给定大小 (107 p-1·s-1·cm-2·sr-1),动物将被随机纳入载体对照或使用指定剂量的 Lorlatinib 进行 3、7 或 14 天治疗。药物通过皮下植入的 Alzet 渗透泵给药。治疗后,处死小鼠,对 GBM 肿瘤进行显微解剖,并将组织在液氮中快速冷冻。剩余的大脑进行组织学处理。在大鼠中,PF-06463922 显示出低血浆清除率、中等分布体积、合理的半衰期、p-糖蛋白 1 介导的外排倾向低以及 100% 的生物利用度。在体内,PF-06463922 通过抑制 ROS1 磷酸化和下游信号分子,以及抑制肿瘤中的细胞周期蛋白 Cyclin D1,在表达人 CD74-ROS1 和 Fig-ROS1 的 NIH3T3 异种移植模型中显示出细胞减灭性抗肿瘤功效。在体内,PF-06463922还在携带表达EML4-ALK、EML4-ALK-L1196M、EML4-ALK-G1269A、EML4-ALK-G1202R或NPM-ALK的肿瘤异种移植物的小鼠中表现出显着的抗肿瘤活性。
|
| 酶活实验 |
微流体迁移率变动测定用于测量重组人野生型和突变型 ALK 激酶结构域蛋白(氨基酸 1093-1411)中的激酶活性,这些蛋白是通过杆状病毒表达和 MgATP 自磷酸化内部产生的。反应含有 3 μM 5-FAM-KKSRGDYMTMQIG-CONH2)、5 mM MgCl2、1.3 nM 野生型 ALK 或 0.5 nM 突变型 ALK(适合在反应 1 小时后产生 15-20% 的肽底物磷酸化),以及25 mM Hepes(pH 7.1)中的 ATP 浓度(Km)。动力学和晶体学研究的结果表明该抑制剂具有 ATP 竞争性。将转化率 (%) 拟合到竞争性抑制方程即可得出 Ki 值。检测 ROS1 酶的程序与检测 ALK 的程序相同,不同之处在于使用 0.25 nM 重组人 ROS1 催化结构域(氨基酸 1883-2347)。 206 激酶组用于评估激酶抑制剂的选择性。
|
| 细胞实验 |
在 96 孔板中,将细胞接种在含有 10% FBS 的生长培养基中,并在 37°C 下孵育整晚。第二天将连续稀释的 Lorlatinib 或合适的对照添加到指定的孔中后,将细胞在 37°C 下孵育 72 小时。为了确定相对细胞数,进行了 CellTiter-Glo 测定。使用四参数分析方法来拟合浓度-响应曲线并确定IC50值。
|
| 动物实验 |
In LSL-FIG-ROS1;Cdkn2a−/−;LSL-Luc mice, de novoGBM tumorigenesis is induced by intracranial stereotactic injections of Adeno-Cre, as previously reported. BLI is used to track the development of tumors as will be discussed below. Animals are randomly assigned to either vehicle control or 3-, 7-, or 14-day treatments with the prescribed doses of lerlatinib once tumors reach a specific size (107 p -1·s -1·cm -2·sr -1). The medication is delivered via s.c. implanted Alzet osmotic pumps. Following therapy, GBM tumors are microdissected, tissues are flash-frozen in liquid N2, and mice are killed. For histology, the remaining brains are processed.
|
| 参考文献 |
J Med Chem.2014 Jun 12;57(11):4720-44;Clin Cancer Res.2012 Sep 1;18(17):4570-9.
|
| 其他信息 |
Although crizotinib demonstrates robust efficacy in anaplastic lymphoma kinase (ALK)-positive non-small-cell lung carcinoma patients, progression during treatment eventually develops. Resistant patient samples revealed a variety of point mutations in the kinase domain of ALK, including the L1196M gatekeeper mutation. In addition, some patients progress due to cancer metastasis in the brain. Using structure-based drug design, lipophilic efficiency, and physical-property-based optimization, highly potent macrocyclic ALK inhibitors were prepared with good absorption, distribution, metabolism, and excretion (ADME), low propensity for p-glycoprotein 1-mediated efflux, and good passive permeability. These structurally unusual macrocyclic inhibitors were potent against wild-type ALK and clinically reported ALK kinase domain mutations. Significant synthetic challenges were overcome, utilizing novel transformations to enable the use of these macrocycles in drug discovery paradigms. This work led to the discovery of 8k (PF-06463922), combining broad-spectrum potency, central nervous system ADME, and a high degree of kinase selectivity.[1]
The oncogenic ROS1 gene fusion (Fig-ROS1) was first identified in glioblastoma cells over two decades ago. Recently, ROS1 gene rearrangements were further discovered in a variety of human cancers, including lung adenocarcinoma, cholangiocarcinoma, ovarian cancer, gastric adenocarcinoma, colorectal cancer, inflammatory myofibroblastic tumor, angiosarcoma, and epithelioid hemangioendothelioma, providing additional evidence for ROS1 as an attractive cancer target. The 1st generation Met/ALK/ROS1 inhibitor XALKORI ® (crizotinib) has demonstrated promising clinical response in ROS1 fusion positive NSCLC. But similar to what was seen with acquired ALK secondary resistant mutations in XALKORI refractory patients, a ROS1 kinase domain mutant–ROS1G2032R has been identified in a ROS1 positive NSCLC patient who developed resistance to XALKORI. Therefore, there is an urgent need to develop agents that can overcome this type of resistance. PF-06463922 is a novel, orally available, ATP-competitive small molecule inhibitor of ROS1/ALK with exquisite potency against ROS1 kinase. PF-06463922 inhibited the catalytic activity of recombinant ROS1 with a mean Ki of < 0.005 nM, and inhibited ROS1 autophosphorylation at IC50 values ranging from 0.1 nM to 1 nM cross a panel of cell lines harboring oncogenic ROS1 fusion variants including CD74-ROS1, SLC34A2-ROS1 and Fig-ROS1. PF-06463922 also inhibited cell proliferation and induced cell apoptosis at sub- to low-nanomolar concentrations in the HCC78 human NSCLC cells harboring SLC34A2-ROS1 fusions and the BaF3-CD74-ROS1 cells expressing human CD74-ROS1. In the BaF3 cells engineered to express the XALKORI resistant CD74-ROS1G2032R mutant, PF-06463922 demonstrated nanomolar potency against either ROS1G2032R cellular activity or cell proliferation. In vivo, PF-06463922 demonstrated marked cytoreductive antitumor efficacy at low nanomolar concentration in the NIH3T3 xenograft models expressing human CD74-ROS1 and Fig-ROS1. The antitumor efficacy of PF-06463922 was dose dependent and strongly correlated to inhibition in ROS1 phosphorylation and the downstream signaling molecules pSHP1, pSHP2 and pErk1/2, as well as inhibition of the cell cycle protein Cyclin D1 in tumors. To our knowledge, PF-06463922 is the first reported ROS1 inhibitor that is capable of blocking the resistant ROS1G2032R mutant at predicted pharmacologically relevant concentrations. Our data indicate that PF-06463922 has great potential for treating ROS1 fusion positive cancers including those from patients who relapsed from XALKORI therapy due to acquired ROS1G2032Rmutation.[2] |
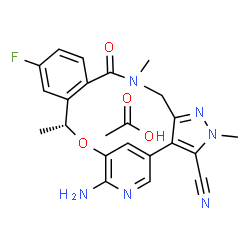
| 分子式 |
C23H23FN6O4
|
|---|---|
| 分子量 |
466.464927911758
|
| 精确质量 |
466.176
|
| CAS号 |
1924207-18-0
|
| 相关CAS号 |
2135926-03-1;2306217-6 (hydrate) ;1924207-18-0 (PF-06463922 acetate); 1454846-35-5;
|
| PubChem CID |
124203822
|
| 外观&性状 |
Typically exists as solid at room temperature
|
| tPSA |
147Ų
|
| 氢键供体(HBD)数目 |
2
|
| 氢键受体(HBA)数目 |
9
|
| 可旋转键数目(RBC) |
0
|
| 重原子数目 |
34
|
| 分子复杂度/Complexity |
731
|
| 定义原子立体中心数目 |
1
|
| SMILES |
FC1C=CC2C(N(C)CC3C(=C(C#N)N(C)N=3)C3=CN=C(C(=C3)O[C@H](C)C=2C=1)N)=O.OC(C)=O
|
| InChi Key |
BLNAIBLTPYGILH-RFVHGSKJSA-N
|
| InChi Code |
InChI=1S/C21H19FN6O2.C2H4O2/c1-11-15-7-13(22)4-5-14(15)21(29)27(2)10-16-19(17(8-23)28(3)26-16)12-6-18(30-11)20(24)25-9-12;1-2(3)4/h4-7,9,11H,10H2,1-3H3,(H2,24,25);1H3,(H,3,4)/t11-;/m1./s1
|
| 化学名 |
acetic acid;(16R)-19-amino-13-fluoro-4,8,16-trimethyl-9-oxo-17-oxa-4,5,8,20-tetrazatetracyclo[16.3.1.02,6.010,15]docosa-1(22),2,5,10(15),11,13,18,20-octaene-3-carbonitrile
|
| 别名 |
Lorlatinib acetate; PF-06463922 acetate; 1924207-18-0; TE9WI16FEU; 2H-4,8-Methenopyrazolo(4,3-H)(2,5,11)benzoxadiazacyclotetradecine-3-carbonitrile, 7-amino-12-fluoro-10,15,16,17-tetrahydro-2,10,16-trimethyl-15-oxo-, (10R)-, acetate; acetic acid;(16R)-19-amino-13-fluoro-4,8,16-trimethyl-9-oxo-17-oxa-4,5,8,20-tetrazatetracyclo[16.3.1.02,6.010,15]docosa-1(22),2,5,10(15),11,13,18,20-octaene-3-carbonitrile; UNII-TE9WI16FEU; PF 06463922 acetate; .
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples
|
|---|---|
| 溶解度 (体内实验) |
注意: 如下所列的是一些常用的体内动物实验溶解配方,主要用于溶解难溶或不溶于水的产品(水溶度<1 mg/mL)。 建议您先取少量样品进行尝试,如该配方可行,再根据实验需求增加样品量。
注射用配方
注射用配方1: DMSO : Tween 80: Saline = 10 : 5 : 85 (如: 100 μL DMSO → 50 μL Tween 80 → 850 μL Saline)(IP/IV/IM/SC等) *生理盐水/Saline的制备:将0.9g氯化钠/NaCl溶解在100 mL ddH ₂ O中,得到澄清溶液。 注射用配方 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (如: 100 μL DMSO → 400 μL PEG300 → 50 μL Tween 80 → 450 μL Saline) 注射用配方 3: DMSO : Corn oil = 10 : 90 (如: 100 μL DMSO → 900 μL Corn oil) 示例: 以注射用配方 3 (DMSO : Corn oil = 10 : 90) 为例说明, 如果要配制 1 mL 2.5 mg/mL的工作液, 您可以取 100 μL 25 mg/mL 澄清的 DMSO 储备液,加到 900 μL Corn oil/玉米油中, 混合均匀。 View More
注射用配方 4: DMSO : 20% SBE-β-CD in Saline = 10 : 90 [如:100 μL DMSO → 900 μL (20% SBE-β-CD in Saline)] 口服配方
口服配方 1: 悬浮于0.5% CMC Na (羧甲基纤维素钠) 口服配方 2: 悬浮于0.5% Carboxymethyl cellulose (羧甲基纤维素) 示例: 以口服配方 1 (悬浮于 0.5% CMC Na)为例说明, 如果要配制 100 mL 2.5 mg/mL 的工作液, 您可以先取0.5g CMC Na并将其溶解于100mL ddH2O中,得到0.5%CMC-Na澄清溶液;然后将250 mg待测化合物加到100 mL前述 0.5%CMC Na溶液中,得到悬浮液。 View More
口服配方 3: 溶解于 PEG400 (聚乙二醇400) 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.1438 mL | 10.7190 mL | 21.4381 mL | |
| 5 mM | 0.4288 mL | 2.1438 mL | 4.2876 mL | |
| 10 mM | 0.2144 mL | 1.0719 mL | 2.1438 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1


































