| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 10 mM * 1 mL in DMSO |
|
||
| 5mg |
|
||
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| 500mg |
|
||
| 1g |
|
||
| Other Sizes |
|
| 靶点 |
c-Met (Ki = 4.8 nM)
|
|---|---|
| 体外研究 (In Vitro) |
体外活性:PF-04217903 比星形孢菌素或 PF-02341066 更具选择性,对 208 种激酶组中的 c-Met 的选择性高出 1000 倍以上,但与 PF-04217903 相比,它更容易受到 c-Met 致癌突变的影响,从而减弱效力。 02341066。除了 WT c-Met 之外,PF-04217903 还表现出类似的抑制 c-Met-H1094R、c-Met-R988C 和 c-Met-T1010I 活性的效力,IC50 分别为 3.1 nM、6.4 nM 和 6.7 nM,分别,但对 c-Met-Y1230C 没有抑制活性,IC50 > 10 μM。 PF-04217903 与舒尼替尼联合显着抑制内皮细胞,但不抑制肿瘤细胞 B16F1、Tib6、EL4 和 LLC PF-04217903 显着抑制 LXFA 526L 和 LXFA 1647L 的克隆生长,IC50 值为 16 nM 和 13 nM,分别与西妥昔单抗联合使用时产生相加效应。 PF-04217903 有效抑制 c-Met 驱动的过程,例如各种肿瘤细胞的细胞生长、运动、侵袭和形态。 PF-04217903 处理 (2 μM) 会增加 GTL-16 细胞的细胞死亡,这涉及磷酸化 4E-BP1、ERK/MAPK 相关蛋白和 PI3K/AKT 通路的下调。激酶测定:将含有内源性人WT c-Met的A549细胞接种到96孔板的生长培养基中并培养过夜。在测定的第二天,将生长培养基更换为无血清培养基(含 0.04% BSA)。将 PF-04217903 的系列稀释液添加到每个孔中,并将细胞在 37°C 下孵育 1 小时。然后将 40 ng/mL HGF 添加到细胞中 20 分钟。用补充有 1 mM Na3VO4 的 HBSS 洗涤细胞一次,并使用裂解缓冲液从细胞中产生蛋白质裂解物。 c-Met 的磷酸化通过 ELISA 方法进行评估,该方法利用 c-Met 特异性的捕获抗体和磷酸化酪氨酸残基特异性的检测抗体。将抗体包被的板在蛋白质裂解物存在下于 4 °C 孵育过夜,并用 1% Tween 20 的 PBS 溶液清洗七次。 HRP-PY20(辣根过氧化物酶缀合的抗磷酸酪氨酸)在封闭缓冲液中按 1:500 稀释,并添加到每个板中 30 分钟。然后再次洗涤板,并添加 TMB 过氧化物酶底物以启动 HRP 依赖性比色反应,并通过添加 0.09 N H2SO4 终止反应。 ELISA 终点是使用分光光度计在 450 nm 处测量的吸光度。 IC50 值是利用基于 Microsoft Excel 的四参数分析方法通过浓度-响应曲线拟合来计算的。细胞测定:用不同浓度的 PF-04217903 处理细胞(B16F1、Tib6、EL4 和 LLC、HUVEC 和 C166 细胞)4 天。通过使用库尔特计数器对每个孔的内容物进行计数来评估细胞增殖。
|
| 体内研究 (In Vivo) |
尽管无法抑制舒尼替尼敏感的 B16F1 和 Tib6 肿瘤模型中的肿瘤生长,但与单独使用舒尼替尼或 PF-04217903 相比,PF-04217903 和舒尼替尼的组合可显着抑制舒尼替尼耐药的 EL4 和 LLC 肿瘤模型中的肿瘤生长。血管扩张,表明 HGF/c-Met 轴在舒尼替尼耐药肿瘤中发挥功能作用。
|
| 酶活实验 |
在 96 孔板中,将表达内源性人 WT c-Met 的 A549 细胞置于生长培养基中并生长整夜。实验第二天将生长培养基更换为无血清培养基(含 0.04% BSA)。每个孔接受 PF-04217903 的系列稀释液,并将细胞在 37°C 下孵育一小时。然后用 40 ng/mL 的 HGF 处理细胞 20 分钟。在补充有 1 mM Na3VO4 的 HBSS 中洗涤细胞一次后,使用裂解缓冲液从细胞中提取蛋白质。使用针对 c-Met 特异性的捕获抗体和针对磷酸化酪氨酸残基特异性的检测抗体的 ELISA 技术来测量 c-Met 的磷酸化。将蛋白质裂解物添加到抗体包被的板中,然后在 4°C 下孵育整夜,然后用 PBS 中的 1% Tween 20 清洗七次。每个板用 1:500 稀释的辣根过氧化物酶缀合的抗磷酸酪氨酸 (HRP-PY20) 处理 30 分钟。又一轮清洗板后,通过添加 TMB 过氧化物酶底物启动 HRP 依赖性比色反应,并通过添加 0.09 N H2SO4 停止。使用分光光度计,450 nm 处的吸光度用于确定 ELISA 终点。通过基于Microsoft Excel的四参数分析方法拟合浓度-响应曲线,确定IC50值。
|
| 细胞实验 |
将细胞暴露于不同浓度的 PF-04217903 四天。使用库尔特计数器对每个孔的内容物进行计数,评估细胞增殖。
简而言之,将GTL-16细胞以每孔20000个细胞的速度铺在96孔板上,并用0.5、1或5μMPF-04217903处理。根据需要每3-5天补充一次化合物。细胞在药物存在下生长约4个月。PF-04217903的浓度每月以0.5μM的增量逐渐增加一次,最终浓度为2.5μM。在2.5μMPF-04217903中存活的细胞被扩增和亚克隆。由于其圆形表型,这些抗性细胞被称为R3克隆。 将亲本GTL16和R3细胞接种在添加了10%FBS的RPMI中的150 cm培养皿中,并在5%CO2的加湿气氛中保持在37°C。在70%融合时,GTL16细胞在RPMI/0.1%FBS中饥饿过夜。第二天,每个板在37°C下用DMSO对照或2μMPF-04217903处理6或24小时。用与抑制剂混合的改良RIPA缓冲液(150 mM NACl,50 mM Tris-HCl,pH 7.4;1%NP-40,0.25%脱氧胆酸钠,1 mM EDTA)裂解细胞,并在冰上孵育30分钟。通过5-8秒脉冲的超声波处理完成裂解。细胞裂解物在15000×g下离心20分钟(4°C)以去除细胞碎片。在将样品储存在-80°C下直至磷蛋白富集之前,通过BCA测定上清液的蛋白质产量[4]。 细胞系,包括B16F1、Tib6、EL4和LLC,以及内皮细胞、HUVEC和C166,在24孔组织培养处理板的每个孔中接种104个细胞。如前所述,细胞在标准培养基中生长。细胞用不同浓度(2、0.2和0.02μmol/L)的舒尼替尼、PF-04217903和两种化合物的组合处理4天。通过在库尔特计数机 中计数细胞来测量化合物的功效。使用3种不同浓度(10、100和200 ng/mL)的每种配体,采用类似的方法评估HGF或VEGF对细胞增殖的作用[2]。 |
| 动物实验 |
Immunodeficient nude mice (nu/nu) subcutaneously implanted with tumor cell lines B16F1, EL4, LLC, or Tib6
45 mg/kg Orally Nude mice were maintained under guidelines provided by the Pfizer IACUC. All the tumor cell lines (B16F1, EL4, LLC, and Tib6) in the current study were obtained from American Tissue Culture Collection and were cultured in RPMI 1640 supplemented with glutamine (2 mmol/L) and fetal bovine serum (FBS; 10%). All the cell lines in the current study were authenticated by the supplier. For implantation, tumor cells (1 × 106 cells per mouse) were resuspended in 100 μL of media and 100 μL of matrigel growth factor reduced and were subcutaneously implanted in one of the flanking areas. Tumor-bearing mice were treated once daily with sunitinib malate at 80 mg/kg or PF-04217903 (45 mg/kg) or the combination of both compounds, using oral route of administration. Tumors volumes were assessed using caliper measurement as described. HUVECs and C166 cells were purchased from Lonza Inc. and ATCC, respectively. For in vitro assays, HUVECs were grown in EBM2 media supplemented with a cocktail of growth factors provided by the supplier, and C166 were grown in DMEM supplemented with FBS (10%). |
| 参考文献 | |
| 其他信息 |
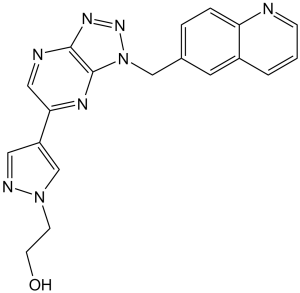
2-[4-[3-(6-quinolinylmethyl)-5-triazolo[4,5-b]pyrazinyl]-1-pyrazolyl]ethanol is a member of quinolines.
PF-04217903 has been used in trials studying the treatment of Neoplasms. MET Tyrosine Kinase Inhibitor PF-04217903 is an orally bioavailabe, small-molecule tyrosine kinase inhibitor with potential antineoplastic activity. MET tyrosine kinase inhibitor PF-04217903 selectively binds to and inhibits c-Met, disrupting the c-Met signaling pathway, which may result in the inhibition of tumor cell growth, migration and invasion of tumor cells, and the induction of death in tumor cells expressing c-Met. The receptor tyrosine kinase c-Met, also known as hepatocyte growth factor (HGF) receptor, is overexpressed or mutated in many tumor cell types, playing an important role in tumor cell proliferation, survival, invasion, and metastasis and angiogenesis. The c-Met receptor tyrosine kinase (RTK) is a key regulator in cancer, in part, through oncogenic mutations. Eight clinically relevant mutants were characterized by biochemical, biophysical, and cellular methods. The c-Met catalytic domain was highly active in the unphosphorylated state (k(cat) = 1.0 s(-1)) and achieved 160-fold enhanced catalytic efficiency (k(cat)/K(m)) upon activation to 425000 s(-1) M(-1). c-Met mutants had 2-10-fold higher basal enzymatic activity (k(cat)) but achieved maximal activities similar to those of wild-type c-Met, except for Y1235D, which underwent a reduction in maximal activity. Small enhancements of basal activity were shown to have profound effects on the acquisition of full enzymatic activity achieved through accelerating rates of autophosphorylation. Biophysical analysis of c-Met mutants revealed minimal melting temperature differences indicating that the mutations did not alter protein stability. A model of RTK activation is proposed to describe how a RTK response may be matched to a biological context through enzymatic properties. Two c-Met clinical candidates from aminopyridine and triazolopyrazine chemical series (PF-02341066 and PF-04217903) were studied. Biochemically, each series produced molecules that are highly selective against a large panel of kinases, with PF-04217903 (>1000-fold selective relative to 208 kinases) being more selective than PF-02341066. Although these prototype inhibitors have similar potencies against wild-type c-Met (K(i) = 6-7 nM), significant differences in potency were observed for clinically relevant mutations evaluated in both biochemical and cellular contexts. In particular, PF-02341066 was 180-fold more active against the Y1230C mutant c-Met than PF-04217903. These highly optimized inhibitors indicate that for kinases susceptible to active site mutations, inhibitor design may need to balance overall kinase selectivity with the ability to inhibit multiple mutant forms of the kinase (penetrance).[1] Molecular and cellular mechanisms underlying resistance/low responsiveness to antiangiogenic compounds are under extensive investigations. Both populations of tumor and stroma (nontumor compartment) seem to contribute in inherent/acquired resistance to antiangiogenic therapy. Here, investigating in vivo efficacy of sunitinib in experimental models resulted in the identification of tumors that were resistant/sensitive to the therapy. Analysis of tumor protein lysates indicated a greater concentration of hepatocyte growth factor (HGF) in resistant tumors than in sensitive ones. In addition, using flow cytometry, c-Met expression was found to be significantly higher in endothelial cells than in tumor cells, suggesting that HGF might target the vascular endothelial cells in resistant tumors. Combination of sunitinib and a selective c-Met inhibitor significantly inhibited tumor growth compared with sunitinib or c-Met inhibitor alone in resistant tumors. Histology and in vitro analyses suggested that combination treatment mainly targeted the vasculature in the resistant tumors. Conversely, systemic injection of HGF in the sensitive tumor models conferred resistance to sunitinib through maintenance of tumor angiogenesis. In conclusion, our study indicates a role for HGF/c-Met pathway in development of resistance to antiangiogenic therapy and suggests a potential strategy to circumvent resistance to vascular endothelial growth factor receptor tyrosine kinase inhibitor in the clinic.[2] Cetuximab (Erbitux®) targets the epidermal growth factor receptor (EGFR) and is approved for treatment of colorectal and head and neck cancer. Despite wide expression of EGFR, only a subgroup of cancer patients responds to cetuximab therapy. In the present study we assessed the cetuximab response in vivo of 79 human patient-derived xenografts originating from five tumour histotypes. We analysed basic tumour characteristics including EGFR expression and activation, mutational status of KRAS, BRAF and NRAS, the expression of EGFR ligands and the activation of HER3 (ErbB3) and the hepatocyte growth factor receptor MET. Based on these results, a cetuximab response score including positive and negative factors affecting therapeutic response is proposed. Positive factors are high expression and activation of EGFR and its ligands epiregulin or amphiregulin, negative factors are markers for downstream pathway activation independent of EGFR. In cetuximab resistant NSCL adenocarcinoma LXFA 526 and LXFA 1647, overexpression due to gene amplification and strong activation of MET was identified. Knock-down of MET by siRNA in the corresponding cell lines showed that anchorage-independent growth and migration are dependent on MET. MET knock down sensitized LXFA 526L and LXFA 1647L to EGF. Combined treatments of a MET inhibitor and cetuximab were additive. Therefore, combination therapy of cetuximab and a MET inhibitor in selected lung cancer patients could be of high clinical significance.[3] In recent years, there have been notable advances with the development of anticancer drugs including those targeting protein tyrosine kinases such as the c-Met receptor, which has been implicated in the development and progression of several cancers. However, despite such progress, drug resistance continues to be the single most important cause of cancer treatment failure, and understanding the mechanisms of drug resistance remains a major hurdle in treating patients with recurrent disease. PF-04217903 is a small-molecule c-Met kinase inhibitor that potently inhibits c-Met-driven processes such as cell growth (proliferation and survival), motility, invasion, and morphology of a variety of tumor cells. Resistance to PF-04217903 was observed in GTL-16, a gastric carcinoma cell line with a constitutively activated c-Met receptor. In this report, mass spectrometry (MS) based quantitative phosphoproteomic analysis was used to determine changes in signaling pathways in the parental cells in response to c-Met inhibition and to investigate the changes in protein levels and related canonical pathways in both parental and PF-04217903 resistant (R3) clones in response to c-Met inhibition. The quantitative MS workflow included phosphoprotein enrichment of cell lysates from six treatment conditions: in-solution digestion, chemical labeling of peptides with a set of 6-plex isobaric tandem mass tags (TMT), HILIC fractionation, phosphopeptide enrichment, and nano LC-MS/MS on a LTQ-Orbitrap mass spectrometer. An investigation of these quantitative datasets using Ingenuity Pathways Analysis (IPA) revealed pathway changes in the various treatments that were consistent with previously observed transcriptomic and phenotypic changes. Proteomic analysis also revealed an increase in B-Raf expression in R3 clones. Expression profiling confirmed that B-Raf gene copy number was up-regulated and also indicated the presence of a mutated form of B-Raf. Using a bottom-up MS approach, SND-1 was identified as the B-Raf fusion partner. The discovery of this novel B-Raf fusion protein presents a novel target with potential clinical implications in the treatment of patients resistant to c-Met inhibitors.[4] |
| 分子式 |
C19H16N8O
|
|---|---|
| 分子量 |
372.38
|
| 精确质量 |
372.144
|
| 元素分析 |
C, 61.28; H, 4.33; N, 30.09; O, 4.30
|
| CAS号 |
956905-27-4
|
| 相关CAS号 |
PF-04217903 mesylate;956906-93-7;PF-04217903 phenolsulfonate;1159490-85-3; 956905-27-4; 1159490-83-1 (monophosphate)
|
| PubChem CID |
17754438
|
| 外观&性状 |
white solid powder
|
| 密度 |
1.5±0.1 g/cm3
|
| 沸点 |
718.1±60.0 °C at 760 mmHg
|
| 闪点 |
388.1±32.9 °C
|
| 蒸汽压 |
0.0±2.4 mmHg at 25°C
|
| 折射率 |
1.807
|
| LogP |
0.3
|
| tPSA |
107.43
|
| 氢键供体(HBD)数目 |
1
|
| 氢键受体(HBA)数目 |
7
|
| 可旋转键数目(RBC) |
5
|
| 重原子数目 |
28
|
| 分子复杂度/Complexity |
524
|
| 定义原子立体中心数目 |
0
|
| SMILES |
OCCN1C=C(C2C=NC3N=NN(C=3N=2)CC2C=C3C(N=CC=C3)=CC=2)C=N1
|
| InChi Key |
PDMUGYOXRHVNMO-UHFFFAOYSA-N
|
| InChi Code |
InChI=1S/C19H16N8O/c28-7-6-26-12-15(9-22-26)17-10-21-18-19(23-17)27(25-24-18)11-13-3-4-16-14(8-13)2-1-5-20-16/h1-5,8-10,12,28H,6-7,11H2
|
| 化学名 |
2-[4-[3-(quinolin-6-ylmethyl)triazolo[4,5-b]pyrazin-5-yl]pyrazol-1-yl]ethanol
|
| 别名 |
PF04217903; PF4217903; PF-4217903; PF 04217903; 2-(4-(1-(quinolin-6-ylmethyl)-1H-[1,2,3]triazolo[4,5-b]pyrazin-6-yl)-1H-pyrazol-1-yl)ethanol; PF-4217903; UNII-CYJ9ATV1IJ; Met tyrosine kinase inhibitor PF-04217903; PF-04217903; PF 4217903
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
|
|||
|---|---|---|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: 2 mg/mL (5.37 mM) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 悬浮液;超声助溶。
例如,若需制备1 mL的工作液,可将100 μL 20.0 mg/mL澄清DMSO储备液加入400 μL PEG300中,混匀;然后向上述溶液中加入50 μL Tween-80,混匀;加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: 2 mg/mL (5.37 mM) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 悬浊液; 超声助溶。 例如,若需制备1 mL的工作液,可将 100 μL 20.0mg/mL澄清的DMSO储备液加入到900μL 20%SBE-β-CD生理盐水中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 View More
配方 3 中的溶解度: ≥ 2 mg/mL (5.37 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 配方 4 中的溶解度: 1% DMSO+30% polyethylene glycol+1% Tween 80: 30 mg/mL 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.6854 mL | 13.4271 mL | 26.8543 mL | |
| 5 mM | 0.5371 mL | 2.6854 mL | 5.3709 mL | |
| 10 mM | 0.2685 mL | 1.3427 mL | 2.6854 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
| NCT Number | Recruitment | interventions | Conditions | Sponsor/Collaborators | Start Date | Phases |
| NCT00706355 | Terminated | Drug: PF-04217903 | Neoplasms | Pfizer | August 2008 | Phase 1 |
Endothelial cells, but not tumor cells, are mainly targeted by HGF/c-Met axis.Cancer Res.2010Dec 15;70(24):10090-100. |
Combination of sunitinib and PF-04217903 has additive effect compared with sunitinib monotherapy. Efficacy of combination treatment (sunitinib plus PF-04217903) in sensitive or resistant tumors.Cancer Res.2010Dec 15;70(24):10090-100. |
Inhibition of angiogenesis is one of the mechanisms by which combination treatment affects tumor growth.Cancer Res.2010Dec 15;70(24):10090-100. |
 Umikibart
Umikibart
 MET-IN-1
MET-IN-1
 EMD 1204831
EMD 1204831
 KRC-00715
KRC-00715
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1






























 COA
COA



