| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 10 mM * 1 mL in DMSO |
|
||
| 1mg |
|
||
| 5mg |
|
||
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| Other Sizes |
|
| 靶点 |
c-Kit (IC50 = 10 nM); cFMS (IC50 = 20 nM); FLT3 (IC50 = 160 nM); KDR (IC50 = 350 nM); LCK (IC50 = 860 nM); FLT1 (IC50 = 880 nM); NTRK3 (IC50 = 890 nM)
|
|---|---|
| 体外研究 (In Vitro) |
体外活性:在M-NFS-60、Bac1.2F5和M-07e细胞中,Pexidartinib抑制CSF1依赖性增殖,IC50分别为0.44 μM、0.22 μM和0.1 μM。激酶检测:Pexidartinib (PLX-3397) 是一种有效、选择性和 ATP 竞争性 CSF1R (cFMS) 和 c-Kit 抑制剂,对 c-Kit 和 CSF1R 的选择性比其他相关激酶(例如 FLT3)高 10 至 100 倍、KDR (VEGFR2)、LCK、FLT1 (VEGFR1) 和 NTRK3 (TRKC),IC50 分别为 160、350、860、880 和 890 nM。
|
| 体内研究 (In Vivo) |
在 MMTV-PyMT 小鼠中,Pexidartinib(40 mg/kg,口服)通过 CD45+CD11b+Ly6C−Ly6G−F4/80+ 显着抑制稳态和 PTX 诱导的肿瘤浸润。 Pexidartinib/PTX 治疗还导致乳腺肿瘤内 CD31+ 血管密度显着降低,同时诱导细胞凋亡和坏死。在携带 GL261 肿瘤的 C57 小鼠中,Pexidartinib (po) 抑制胶质母细胞瘤侵袭。在 cmo 小鼠中,PLX3397 通过减少尾部和爪子的侵蚀性骨病变以及循环 MIP-1α 的水平,显着减轻自身炎症性疾病。在患有 B16F10 黑色素瘤的小鼠中,Pexidartinib(45 mg/kg,口服)可增强 CD8 介导的黑色素瘤免疫治疗。
|
| 酶活实验 |
Pexidartinib (PLX3397)的生化选择性和效力:[1]
Pexidartinib(PLX3397)选择性抑制c-Fms和c-Kit受体酪氨酸激酶,生化IC50值分别为0.02µM和0.01µM(图S6A)Pexidartinib(PLX3397)通过使用基于支架和X射线结构的发现方法被鉴定为一种有效的CSF-1R和c-KIT激酶抑制剂。在226种不同激酶的全面筛选中,包括所有蛋白激酶亚家族和几种脂质激酶的代表,0.03µM和1.0µM的Pexidartinib (PLX3397)仅显著抑制了其他五种激酶Pexidartinib(PLX3397)是基于抑制小鼠粒细胞白血病细胞系M-NFS-60的CSF1依赖性增殖而选择的,IC50为0.44µM,小鼠巨噬细胞系Bac1.2F5的IC50为0.22µM。依赖添加SCF生长的人急性巨核细胞白血病细胞系M-07e被Pexidartinib (PLX3397)抑制,IC50为0.1µM。这些亚微摩尔效力证实,培昔达替尼(PLX3397)可以进入细胞并抑制Fms驱动的细胞生长。[1] Pexidartinib (PLX-3397) 是一种 ATP 竞争性、强效、选择性的 CSF1R (cFMS) 和 c-Kit 抑制剂,与其他相关激酶、FLT3、KDR (VEGFR2)、LCK、FLT1 相比,对 c-Kit 和 CSF1R 具有选择性(VEGFR1) 和 NTRK3 (TRKC),IC50 值分别为 160、350、860、880 和 890 nM。 |
| 细胞实验 |
增殖、凋亡和集落形成测定[5]
根据制造商的说明,使用Methocult H4230进行菌落形成测定。简言之,将约5000个细胞接种有或没有多西环素(10ng/ml),菌落生长7天,然后用对碘硝基四氮唑氯化物染色。用CellTiter-Glo测定法在72小时内评估增殖。通过膜联蛋白V掺入的流式细胞术分析,结合碘化丙啶或DilC1(5)掺入,对细胞凋亡进行多参数评估。根据制造商说明进行流式细胞术的染色和检测程序。 |
| 动物实验 |
Neonatal mice
0.25, 1 mg/kg I.P. twice daily for 8 days Two murine models of mammary tumor development were used to analyze response to chemotherapy (Supplementary Fig. S3). The first model used MMTV-PyMT mice (Supplementary Fig. S3A). The 80-day-old MMTV-PyMT female littermates were randomized by initial tumor volume and fed either PLX3397 (20, 61, 62) formulated in mouse chow or control chow (provided by Plexxikon Inc). PLX3397 was formulated in mouse chow so that the average dose per animal per day was 40 mg/kg. When PLX3397-treated MMTV-PyMT mice reached 85 days of age, they were then administered PTX (Hospira) every 5 days by i.v. injection into the retroorbital plexus. PTX was given at 10 mg/kg of the animal per injection, diluted in PBS. Tumor burden was evaluated by caliper measurement every 5 days following the start of PLX3397 treatment. Prior to tissue collection, mice were cardiac-perfused with PBS to clear peripheral blood. Mammary tumor tissue from PBS-perfused MMTV-PyMT mice was analyzed by flow cytometry and qRT-PCR 2 days after the second dose of PTX, when metastatic burden and tumor grade were determined. Primary tumor burden was determined by caliper measurements on live sedated mice. Metastatic burden was assessed by serial sectioning of formalin-fixed paraffin-embedded lung tissue whereby the entire lung was sectioned and the number of metastatic foci (>5 cells) was determined on 6 sections taken every 100 µm following H&E staining. Lungs from >10 mice/group were analyzed[1]. A Ten-week-old mice were fed a chow or high-fat diet for 10 weeks and then treated with PLX3397 via oral gavage (50 mg/kg) every second day for 3 weeks, with subsequent monitoring of glucose tolerance, insulin sensitivity and assessment of adipose tissue immune cells.PLX3397 treatment substantially reduced macrophage numbers in adipose tissue of both chow and high-fat diet fed mice without affecting total myeloid cell levels. Despite this, PLX3397 did not greatly alter glucose homeostasis, did not affect high-fat diet-induced increases in visceral fat cytokine expression (Il-6 and Tnfa) and had limited effect on the phosphorylation of the stress kinases JNK and ERK and macrophage polarization.[4] |
| 药代性质 (ADME/PK) |
Absorption, Distribution and Excretion
Following administration of single doses in healthy subjects and multiple doses in patients, the mean Cmax was 8625 ng/mL and the mean AUC was 77465 ngxh/mL. The median Tmax was 2.5 hours and the time to reach the steady state was approximately 7 days. Administration of pexidartinib with a high fat meal resulted in an increased drug Cmax and AUC by 100%, with a delay in Tmax by 2.5 hours. Pexidartinib is predominantly excreted via feces, where fecal excretion accounts for 65% of total pexidartinib elimination. Via this route of elimination, about 44% of the compound found in feces is recovered as unchanged parent drug. The renal elimination accounts for 27% of pexidartinib elimination, where more than 10% of the compound is found as the N-glucuronide metabolite. The apparent volume of distribution of pexidartinib is about 187 L. In rats, pexidartinib was shown to penetrate into the central nervous system. The apparent clearance is about 5.1 L/h. Metabolism / Metabolites Pexidartinib primarily undergoes oxidation mediated by hepatic CYP3A4 and glucuronidation by UGT1A4. Following UGT1A4-mediated glucuronidation, a major inactive N-glucuronide metabolite is formed with approximately 10% higher exposure than the parent drug after a single dose administration of pexidartinib. Based on the findings of _in vitro_ studies, CYP1A2 and CYP2C9 may also play a minor role in drug metabolism. Biological Half-Life The elimination half-life is about 26.6 hours. |
| 毒性/毒理 (Toxicokinetics/TK) |
Hepatotoxicity
Elevations in serum aminotransferase levels are common during pexidartinib therapy, occurring in 50% to 90% of patients and rising above 5 times the upper limit of the normal range in 12% to 20%. In addition, elevations in alkaline phosphatase levels occur in up to 20% of treated persons. In registration trials, clinically apparent liver injury with jaundice developed in 5% of patients. The time to onset of liver injury was typically between 2 and 6 weeks, and the pattern of liver enzyme elevations was mixed or cholestatic. Autoimmune and immune-allergic features were not prominent. Liver biopsy demonstrated bile duct injury and loss, and at least 3 patients in studies for conditions other than TGCT developed bile duct paucity and features of vanishing bile duct syndrome that ultimately led to liver transplantation in one subject. Pexidartinib has had limited clinical use and the frequency and spectrum of acute liver injury with its use is not yet well defined. Likelihood score: B (likely cause of clinically apparent liver injury). Effects During Pregnancy and Lactation ◉ Summary of Use during Lactation No information is available on the clinical use of pexidartinib during breastfeeding. Because pexidartinib is over 99% bound to plasma proteins, the amount in milk is likely to be low. However, the manufacturer recommends that breastfeeding be discontinued during pexidartinib therapy and for 1 week after the last dose. ◉ Effects in Breastfed Infants Relevant published information was not found as of the revision date. ◉ Effects on Lactation and Breastmilk Relevant published information was not found as of the revision date. Protein Binding Based on the findings of _in vitro_ plasma protein binding study, pexidartinib is about 99% bound to serum proteins, where it is extensively bound to human serum albumin by 99.9% and alpha-1-acid glycoprotein by 89.9%. |
| 参考文献 |
|
| 其他信息 |
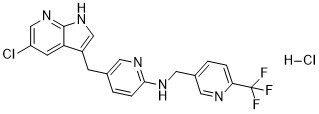
Pexidartinib Hydrochloride is the hydrochloride salt form of pexidartinib, a small-molecule receptor tyrosine kinase (RTK) inhibitor of proto-oncogene receptor tyrosine kinase (KIT), colony-stimulating factor-1 receptor (CSF1R) and FMS-like tyrosine kinase 3 (FLT3), with antineoplastic activity. Upon oral administration, pexidartinib targets, binds to and inhibits phosphorylation of KIT, CSF1R and FLT3 harboring an internal tandem duplication (ITD) mutation. This results in the inhibition of tumor cell proliferation. FLT3, CSF1R and FLT3 are overexpressed or mutated in many cancer cell types and play major roles in tumor cell proliferation and metastasis.
See also: Pexidartinib (has active moiety). Pexidartinib is a pyrrolopyridine that is 5-chloro-1H-pyrrolo[2,3-b]pyridine which is substituted by a [6-({[6-(trifluoromethyl)pyridin-3-yl]methyl}amino)pyridin-3-yl]methyl group at position 3. It is a potent multi-targeted receptor tyrosine kinase inhibitor of CSF-1R, KIT, and FLT3 (IC50 of 20 nM, 10 nM and 160 nM, respectively). Approved by the FDA for the treatment of adult patients with symptomatic tenosynovial giant cell tumor (TGCT). It has a role as an EC 2.7.10.1 (receptor protein-tyrosine kinase) inhibitor and an antineoplastic agent. It is a pyrrolopyridine, an organochlorine compound, an aminopyridine, an organofluorine compound and a secondary amino compound. Pexidartinib is a selective tyrosine kinase inhibitor that works by inhibiting the colony-stimulating factor (CSF1)/CSF1 receptor pathway. Pexidartinib was originally developed by Daiichi Sankyo, Inc. and it was approved by the FDA in August 2019 as the first systemic therapy for adult patients with symptomatic tenosynovial giant cell tumor. Tenosynovial giant cell tumor is a rare form of non-malignant tumor that causes the synovium and tendon sheaths to thicken and overgrow, leading to damage in surrounding joint tissue. Debilitating symptoms often follow with tenosynovial giant cell tumors, along with a risk of significant functional limitations and a reduced quality of life in patients. While surgical resection is a current standard of care for tenosynovial giant cell tumor, there are tumor types where surgeries are deemed clinically ineffective with a high risk of lifetime recurrence. Pexidartinib works by blocking the immune responses that are activated in tenosynovial giant cell tumors. In clinical trials, pexidartinib was shown to promote improvements in patient symptoms and functional outcomes in TGCT. Pexidartinib is available in oral formulations and it is commonly marketed as Turalio. Pexidartinib is a Kinase Inhibitor. The mechanism of action of pexidartinib is as a Kinase Inhibitor, and Tyrosine Kinase Inhibitor, and Colony Stimulating Factor Receptor Type 1 (CSF-1R) Inhibitor, and Cytochrome P450 3A Inducer, and Cytochrome P450 2B6 Inhibitor, and UGT1A1 Inhibitor. Pexidartinib is an orally available small molecule multi-kinase inhibitor that is used as an antineoplastic agent in the treatment of tenosynovial giant cell tumors. Pexidartinib is associated with a high rates of serum aminotransferase and alkaline phosphatase elevations during therapy and has been implicated in several cases of clinically apparent liver injury marked by progressive intrahepatic bile duct injury, some of which resulted in liver transplantation or were fatal. Pexidartinib is a small-molecule receptor tyrosine kinase (RTK) inhibitor of proto-oncogene receptor tyrosine kinase (KIT), colony-stimulating factor-1 receptor (CSF1R) and FMS-like tyrosine kinase 3 (FLT3), with antineoplastic activity. Upon oral administration, pexidartinib targets, binds to and inhibits phosphorylation of KIT, CSF1R and FLT3 harboring an internal tandem duplication (ITD) mutation. This results in the inhibition of tumor cell proliferation. FLT3, CSF1R and FLT3 are overexpressed or mutated in many cancer cell types and play major roles in tumor cell proliferation and metastasis. See also: Pexidartinib Hydrochloride (has salt form). Drug Indication Pexidartinib is indicated for the treatment of adult patients with symptomatic tenosynovial giant cell tumor (TGCT) associated with severe morbidity or functional limitations and not amenable to improvement with surgery. Treatment of tenosynovial giant cell tumour. Treatment of benign soft tissue neoplasms Mechanism of Action Tenosynovial giant cell tumor is a rare, non-malignant neoplasm that causes abnormal growth and damage to the synovium, bursae, or tendon sheaths. Recruitment of immune cells, specifically macrophages, is closely associated with the tumor mass formation in tenosynovial giant cell tumors. Macrophages drive tumor-promoting inflammation and play a central role in every stage of tumor progression. As the most abundant immune cells in the tumor microenvironment of solid tumors, macrophages promote processes that enhance tumor survival, such as angiogenesis, tumor cell invasion, and intravasation at the primary site. They also modulate the immune response to tumors to inhibit tumor clearance and directly engage with tumor cells to activate pro-survival signaling pathways. The recruitment, proliferation, and irreversible differentiation of macrophages are regulated by colony-stimulating factor-1 (CSF-1), which is a cytokine that is often translocated and highly expressed in tenosynovial giant cell tumors. Elevated expression of CSF-1 and CSF-1 receptor (CSF1R) has also been implicated in various models of malignant cancers and tumors. Pexidartinib targets the CSF1/CSF1R pathway as a selective CSF1R inhibitor. It stimulates the autoinhibited state of the CSF1R by interacting with the juxtamembrane region of CSF1R, which is responsible for folding and inactivation of the kinase domain, and preventing the binding of CSF1 and ATP to the region. Without the binding of CSF1 to the receptor, CSF1R cannot undergo ligand-induced autophosphorylation. By inhibiting the CSF1R signaling pathway, pexidartinib works to inhibit tumor cell proliferation and downmodulate cells involved in the disease, such as macrophages. It was also shown to inhibit the CD117 or proto-oncogene receptor tyrosine kinase (cKIT), mutant fms-like tyrosine kinase 3 (FLT3), and platelet-derived growth factor receptor (PDGFR)-β, which are all receptor tyrosine kinases that regulate critical cellular processes such as cell proliferation and survival. Immune-regulated pathways influence multiple aspects of cancer development. In this article we demonstrate that both macrophage abundance and T-cell abundance in breast cancer represent prognostic indicators for recurrence-free and overall survival. We provide evidence that response to chemotherapy is in part regulated by these leukocytes; cytotoxic therapies induce mammary epithelial cells to produce monocyte/macrophage recruitment factors, including colony stimulating factor 1 (CSF1) and interleukin-34, which together enhance CSF1 receptor (CSF1R)-dependent macrophage infiltration. Blockade of macrophage recruitment with CSF1R-signaling antagonists, in combination with paclitaxel, improved survival of mammary tumor-bearing mice by slowing primary tumor development and reducing pulmonary metastasis. These improved aspects of mammary carcinogenesis were accompanied by decreased vessel density and appearance of antitumor immune programs fostering tumor suppression in a CD8+ T-cell-dependent manner. These data provide a rationale for targeting macrophage recruitment/response pathways, notably CSF1R, in combination with cytotoxic therapy, and identification of a breast cancer population likely to benefit from this novel therapeutic approach.[1] |
| 分子式 |
C20H16CL2F3N5
|
|---|---|
| 分子量 |
454.275752067566
|
| 精确质量 |
453.073
|
| 元素分析 |
C, 52.88; H, 3.55; Cl, 15.61; F, 12.55; N, 15.42
|
| CAS号 |
2040295-03-0
|
| 相关CAS号 |
Pexidartinib;1029044-16-3; 2040295-03-0 (HCl); 2169310-71-6 (2HCl)
|
| PubChem CID |
73053710
|
| 外观&性状 |
White to off-white solid powder
|
| tPSA |
66.5Ų
|
| 氢键供体(HBD)数目 |
3
|
| 氢键受体(HBA)数目 |
7
|
| 可旋转键数目(RBC) |
5
|
| 重原子数目 |
30
|
| 分子复杂度/Complexity |
537
|
| 定义原子立体中心数目 |
0
|
| InChi Key |
CJLUYLRKLUYCEK-UHFFFAOYSA-N
|
| InChi Code |
InChI=1S/C20H15ClF3N5.ClH/c21-15-6-16-14(10-28-19(16)29-11-15)5-12-2-4-18(26-7-12)27-9-13-1-3-17(25-8-13)20(22,23)24;/h1-4,6-8,10-11H,5,9H2,(H,26,27)(H,28,29);1H
|
| 化学名 |
5-[(5-chloro-1H-pyrrolo[2,3-b]pyridin-3-yl)methyl]-N-[[6-(trifluoromethyl)pyridin-3-yl]methyl]pyridin-2-amine;hydrochloride
|
| 别名 |
PLX3397; PLX-3397; PLX 3397; YS6WAI3XN7; UNII-YS6WAI3XN7; Pexidartinib (hydrochloride); CML-261; Pexidartinib HCl; PLX3397 HCl; YS6WAI3XN7; CML 261; CML261; FP-113; FP 113; FP113; Pexidartinib HCl; Pexidartinib hydrochloride; Turalio; PLX3397 HCl;
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month 注意: 请将本产品存放在密封且受保护的环境中,避免吸湿/受潮。 |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
DMSO: ~60 mg/mL (~132.1 mM)
H2O: < 0.1 mg/mL |
|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 2.5 mg/mL (5.50 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 25.0 mg/mL澄清DMSO储备液加入到400 μL PEG300中,混匀;然后向上述溶液中加入50 μL Tween-80,混匀;加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: ≥ 2.5 mg/mL (5.50 mM) (饱和度未知) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 25.0 mg/mL澄清DMSO储备液加入900 μL 20% SBE-β-CD生理盐水溶液中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 View More
配方 3 中的溶解度: ≥ 2.5 mg/mL (5.50 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 配方 4 中的溶解度: ≥ 2.5 mg/mL (5.50 mM) (饱和度未知) in 5% DMSO + 40% PEG300 + 5% Tween80 + 50% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 5 中的溶解度: ≥ 2.5 mg/mL (5.50 mM) (饱和度未知) in 5% DMSO + 95% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.2013 mL | 11.0064 mL | 22.0129 mL | |
| 5 mM | 0.4403 mL | 2.2013 mL | 4.4026 mL | |
| 10 mM | 0.2201 mL | 1.1006 mL | 2.2013 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
| NCT Number | Recruitment | interventions | Conditions | Sponsor/Collaborators | Start Date | Phases |
| NCT02371369 | Completed | Drug: Pexidartinib Drug: Placebo |
Pigmented Villonodular Synovitis Tenosynovial Giant Cell Tumor |
Daiichi Sankyo, Inc. | May 11, 2015 | Phase 3 |
Combined PLX3397 and PTX treatment inhibits metastasis in a CD8-dependent manner. Cancer Discov. 2011 Jun 1; 1: 54–67. |
PTX in combination with PLX3397 induces antitumor T-cell response. Cancer Discov. 2011 Jun 1; 1: 54–67. |
|
 Pyrithyldione
Pyrithyldione
 Pimicotinib hydrochloride
Pimicotinib hydrochloride
 Edicotinib hydrochloride
Edicotinib hydrochloride
 Enrupatinib
Enrupatinib
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1






























 COA
COA
")
")
")
