| 规格 | 价格 | |
|---|---|---|
| 500mg | ||
| 1g | ||
| Other Sizes |

| 药代性质 (ADME/PK) |
Absorption, Distribution and Excretion
Racemic milnacipram demonstrates an absolute bioavailability of about 85-90% following oral administration. Maximum concentrations of the racemic agent are reached within 2-4 hours after oral dosing, and steady-state levels are obtained by 36-48 hours. Conversely, the relative bioavailability of levomilnacipram has been documented as 92%. The median time to peak concentration Tmax for levomilnacipram is about 6-8 hours after oral administration. After daily dosing of levomilnacipram 120 mg, the mean Cmax value is 341 ng/mL, and the mean steady-state AUC value is 5196 ng.h/mL. In general, the administration of either racemic milnacipram or levomilnacipram with food does not affect the medication's oral bioavailability. Levomilnacipran and its metabolites are eliminated primarily by renal excretion. Following oral administration of 14C-levomilnacipran solution, approximately 58% of the dose is excreted in urine as unchanged levomilnacipran. N-desethyl levomilnacipran is the major metabolite excreted in the urine and accounted for approximately 18% of the dose. Other identifiable metabolites excreted in the urine are levomilnacipran glucuronide (4%), desethyl levomilnacipran glucuronide (3%), p-hydroxy levomilnacipran glucuronide (1%), and p-hydroxy levomilnacipran (1%). The mean volume of distribution recorded for racemic milnacipran following a single intravenous dose to healthy subjects was approximately 400 L. Alternatively, levomilnacipran is widely distributed with an apparent volume of distribution of 387-473 L. The total plasma clearance determined for milnacipran is approximately 40 L/h. Metabolism / Metabolites It has been determined that levomilnacipran undergoes desethylation and hydroxylation to generate desethyl levomilnacipran and p-hydroxy-levomilnacipran, respectively. Both oxidative metabolites undergo further conjugation with glucuronide to form the conjugate milnacipran carbamoyl-O-glucuronide. The desethylation is catalyzed primarily by CYP3A4 with minor contribution by CYP2C8, 2C19, 2D6, and 2J2. Additionally, it is the general understanding that there is no interconversion between the enantiomers of milnacipran in the body. Biological Half-Life The terminal elimination half-life documented for racemic milnacipran is approximately 6-8 hours, where d-milnacipran has a longer elimination half-life of 8-10 hours compared to that of the l-enantionmer at 4-6 hours. Alternatively, the terminal elimination half-life determined specifically for levomilnacipran formulations is about 12 hours. |
|---|---|
| 毒性/毒理 (Toxicokinetics/TK) |
Effects During Pregnancy and Lactation
◉ Summary of Use during Lactation Amounts of milnacipran in breastmilk are low and would not be expected to cause any adverse effects in breastfed infants. However, until more data become available, milnacipran should be used with caution during breastfeeding, especially while nursing a newborn or preterm infant. ◉ Effects in Breastfed Infants Relevant published information was not found as of the revision date. ◉ Effects on Lactation and Breastmilk Galactorrhea is reported by the manufacturer to be a side effect of milnacipran. One woman who was being treated for depression took an intentional overdose of 950 mg of milnacipran orally. From day 5 to day 15 after the overdose, the patient noted a flow of milk from her left breast. The galactorrhea resolved without treatment. In a study of cases of hyperprolactinemia and its symptoms (e.g., gynecomastia) reported to a French pharmacovigilance center, milnacipran was not found to have an increased risk of causing hyperprolactinemia compared to other drugs. An observational study looked at outcomes of 2859 women who took an antidepressant during the 2 years prior to pregnancy. Compared to women who did not take an antidepressant during pregnancy, mothers who took an antidepressant during all 3 trimesters of pregnancy were 37% less likely to be breastfeeding upon hospital discharge. Mothers who took an antidepressant only during the third trimester were 75% less likely to be breastfeeding at discharge. Those who took an antidepressant only during the first and second trimesters did not have a reduced likelihood of breastfeeding at discharge. The antidepressants used by the mothers were not specified. A retrospective cohort study of hospital electronic medical records from 2001 to 2008 compared women who had been dispensed an antidepressant during late gestation (n = 575) to those who had a psychiatric illness but did not receive an antidepressant (n = 1552) and mothers who did not have a psychiatric diagnosis (n = 30,535). Women who received an antidepressant were 37% less likely to be breastfeeding at discharge than women without a psychiatric diagnosis, but no less likely to be breastfeeding than untreated mothers with a psychiatric diagnosis. None of the mothers were taking milnacipran. In a study of 80,882 Norwegian mother-infant pairs from 1999 to 2008, new postpartum antidepressant use was reported by 392 women and 201 reported that they continued antidepressants from pregnancy. Compared with the unexposed comparison group, late pregnancy antidepressant use was associated with a 7% reduced likelihood of breastfeeding initiation, but with no effect on breastfeeding duration or exclusivity. Compared with the unexposed comparison group, new or restarted antidepressant use was associated with a 63% reduced likelihood of predominant, and a 51% reduced likelihood of any breastfeeding at 6 months, as well as a 2.6-fold increased risk of abrupt breastfeeding discontinuation. Specific antidepressants were not mentioned. Protein Binding The protein binding determined for racemic milnacipran is 13%. Conversely, the plasma protein binding documented for levomilnacipran is 22% over a concentration range of 10 to 1000 ng/mL. |
| 参考文献 | |
| 其他信息 |
Pharmacodynamics
When utilized to treat fibromyalgia, the effect of milnacipran on the QTcF interval in patients was measured in a double-blind placebo-and positive-controlled parallel study in 88 healthy subjects using three to six times the recommended therapeutic dose for fibromyalgia at 600 mg/day. After baseline and placebo adjustment, the maximum mean QTcF change was 8 ms - an increase that is generally not considered to be clinically significant. Conversely, when used for treating major depressive disorder (MDD), non-clinical studies have shown that levomilnacipran binds with high affinity to the norepinephrine (NE) and serotonin (5-HT) transporters (Ki = 71-91 nM and 11 nM respectively at human transporters). Levomilnacipran inhibits the uptake of both NE and 5-HT in vitro and in vivo; preferentially inhibiting reuptake of NE over 5-HT by approximately 2-fold. Levomilnacipran does not directly affect the uptake of dopamine or other neurotransmitters. Levomilnacipran has no significant affinity for serotonergic (5-HT1-7), α- and β-adrenergic, muscarinic (M1-5), histamine (H1-4), dopamine (D1-5), opiate, benzodiazepine, and γ-aminobutyric acid (GABA) receptors in vitro. Levomilnacipran has no significant affinity for Ca++, K+, Na+, and Cl– channels and does not inhibit the activity of human monoamine oxidases (MAO-A and MAO-B) or acetylcholinesterase. Moreover, in ECG studies with levomilnacipran used to treat MDD, although no clinically significant changes in QTcF interval (QTcF=QT/RR0.33) were noted, it appears that the agent can cause increases in heart rate and blood pressure. In particular, it appears that the maximum therapeutic dose of levomilnacipran at 120 mg/day is capable of causing a maximum mean difference in heart rate from placebo of 20.2 bpm and a mean difference in systolic and diastolic blood pressure from placebo ranging from 3.8 to 7.2 mmHg and 6.1 to 8.1 mmHg, respectively. Alternatively, a supratherapeutic dose of 300 mg/day is capable of causing a maximum mean difference in heart rate from placebo of 22.1 bpm and a mean difference in systolic and diastolic blood pressure from placebo ranging from 5.4 to 7.9 mmHg and 7.9 to 10.6 mmHg, respectively. |
| 分子式 |
C15H22N2O
|
|---|---|
| 分子量 |
246.34798
|
| 精确质量 |
246.173
|
| CAS号 |
92623-85-3
|
| 相关CAS号 |
Milnacipran hydrochloride;101152-94-7;Milnacipran ((1S-cis) hydrochloride);175131-60-9;Dextromilnacipran;96847-55-1;Milnacipran-d10 hydrochloride;1217774-40-7
|
| PubChem CID |
65833
|
| 外观&性状 |
Typically exists as solid at room temperature
|
| 密度 |
1.1±0.1 g/cm3
|
| 沸点 |
393.0±21.0 °C at 760 mmHg
|
| 熔点 |
228-228.5ºC
|
| 闪点 |
191.5±22.1 °C
|
| 蒸汽压 |
0.0±0.9 mmHg at 25°C
|
| 折射率 |
1.554
|
| LogP |
1.23
|
| tPSA |
46.33
|
| 氢键供体(HBD)数目 |
1
|
| 氢键受体(HBA)数目 |
2
|
| 可旋转键数目(RBC) |
5
|
| 重原子数目 |
18
|
| 分子复杂度/Complexity |
295
|
| 定义原子立体中心数目 |
2
|
| SMILES |
C([C@@]1(C[C@@H]1CN)C1C=CC=CC=1)(=O)N(CC)CC
|
| InChi Key |
GJJFMKBJSRMPLA-HIFRSBDPSA-N
|
| InChi Code |
InChI=1S/C15H22N2O/c1-3-17(4-2)14(18)15(10-13(15)11-16)12-8-6-5-7-9-12/h5-9,13H,3-4,10-11,16H2,1-2H3/t13-,15+/m1/s1
|
| 化学名 |
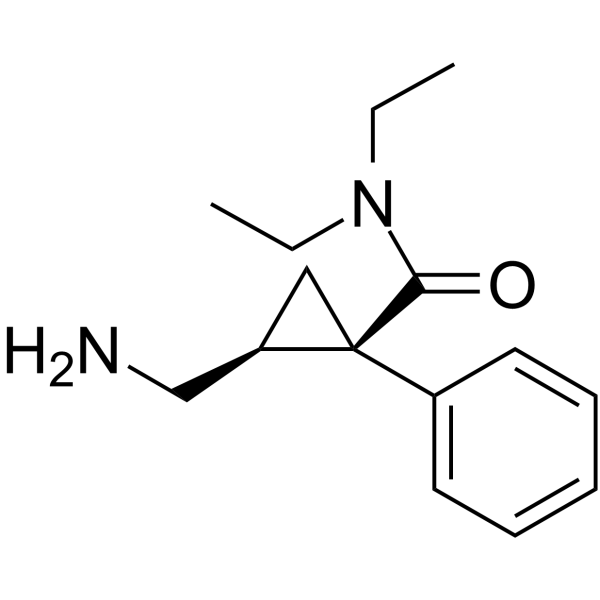
(1R,2S)-2-(aminomethyl)-N,N-diethyl-1-phenylcyclopropane-1-carboxamide
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples
|
|---|---|
| 溶解度 (体内实验) |
注意: 如下所列的是一些常用的体内动物实验溶解配方,主要用于溶解难溶或不溶于水的产品(水溶度<1 mg/mL)。 建议您先取少量样品进行尝试,如该配方可行,再根据实验需求增加样品量。
注射用配方
注射用配方1: DMSO : Tween 80: Saline = 10 : 5 : 85 (如: 100 μL DMSO → 50 μL Tween 80 → 850 μL Saline)(IP/IV/IM/SC等) *生理盐水/Saline的制备:将0.9g氯化钠/NaCl溶解在100 mL ddH ₂ O中,得到澄清溶液。 注射用配方 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (如: 100 μL DMSO → 400 μL PEG300 → 50 μL Tween 80 → 450 μL Saline) 注射用配方 3: DMSO : Corn oil = 10 : 90 (如: 100 μL DMSO → 900 μL Corn oil) 示例: 以注射用配方 3 (DMSO : Corn oil = 10 : 90) 为例说明, 如果要配制 1 mL 2.5 mg/mL的工作液, 您可以取 100 μL 25 mg/mL 澄清的 DMSO 储备液,加到 900 μL Corn oil/玉米油中, 混合均匀。 View More
注射用配方 4: DMSO : 20% SBE-β-CD in Saline = 10 : 90 [如:100 μL DMSO → 900 μL (20% SBE-β-CD in Saline)] 口服配方
口服配方 1: 悬浮于0.5% CMC Na (羧甲基纤维素钠) 口服配方 2: 悬浮于0.5% Carboxymethyl cellulose (羧甲基纤维素) 示例: 以口服配方 1 (悬浮于 0.5% CMC Na)为例说明, 如果要配制 100 mL 2.5 mg/mL 的工作液, 您可以先取0.5g CMC Na并将其溶解于100mL ddH2O中,得到0.5%CMC-Na澄清溶液;然后将250 mg待测化合物加到100 mL前述 0.5%CMC Na溶液中,得到悬浮液。 View More
口服配方 3: 溶解于 PEG400 (聚乙二醇400) 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 4.0593 mL | 20.2963 mL | 40.5927 mL | |
| 5 mM | 0.8119 mL | 4.0593 mL | 8.1185 mL | |
| 10 mM | 0.4059 mL | 2.0296 mL | 4.0593 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1

































