| 规格 | 价格 | |
|---|---|---|
| 500mg | ||
| 1g | ||
| Other Sizes |
| 药代性质 (ADME/PK) |
Absorption, Distribution and Excretion
Rapidly absorbed following oral administration. The peak plasma concentrations of the prodrug, desglymidodrine, is reached about half an hour following drug administration. The metabolites reach their peak plasma concentrations at about 1 to 2 hours following drug administration. The absolute bioavailability of midodrine (measured as desglymidodrine) is 93% and is not affected by food. As desglymidodrine displays poor diffusibility across the blood-brain barrier, it is expected to have minimal effects on the central nervous system. Renal cl=385 mL/minute Renal elimination of midodrine is insignificant. The renal clearance of desglymidodrine is of the order of 385 mL/minute, most, about 80%, by active renal secretion. The actual mechanism of active secretion has not been studied, but it is possible that it occurs by the base-secreting pathway responsible for the secretion of several other drugs that are bases. Midodrine hydrochloride is a prodrug, i.e., the therapeutic effect of orally administered midodrine is due to the major metabolite desglymidodrine, formed by deglycination of midodrine. After oral administration, midodrine hydrochloride is rapidly absorbed. The plasma levels of the prodrug peak after about half an hour and decline with a half-life of approximately 25 minutes, while the metabolite reaches peak blood concentrations about 1 to 2 hours after a dose of midodrine and has a half-life of about 3 to 4 hours. The absolute bioavailability of midodrine (measured as desglymidodrine) is 93%. The bioavailability of desglymidodrine is not affected by food. Approximately the same amount of desglymidodrine is formed after intravenous and oral administration of midodrine. Neither midodrine nor desglymidodrine is bound to plasma proteins to any significant extent. Midodrine is an oral drug for orthostatic hypotension. This drug is almost completely absorbed after oral administration and converted into its active form, 1-(2',5'-dimethoxyphenyl)-2-aminoethanol) (DMAE), by the cleavage of a glycine residue. The intestinal H+-coupled peptide transporter 1 (PEPT1) transports various peptide-like drugs and has been used as a target molecule for improving the intestinal absorption of poorly absorbed drugs through amino acid modifications. Because midodrine meets these requirements, we examined whether midodrine can be a substrate for PEPT1. The uptake of midodrine, but not DMAE, was markedly increased in PEPT1-expressing oocytes compared with water-injected oocytes. Midodrine uptake by Caco-2 cells was saturable and was inhibited by various PEPT1 substrates. Midodrine absorption from the rat intestine was very rapid and was significantly inhibited by the high-affinity PEPT1 substrate cyclacillin, assessed by the alteration of the area under the blood concentration-time curve for 30 min and the maximal concentration. Some amino acid derivatives of DMAE were transported by PEPT1, and their transport was dependent on the amino acids modified. In contrast to neutral substrates, cationic midodrine was taken up extensively at alkaline pH, and this pH profile was reproduced by a 14-state model of PEPT1, which we recently reported. These findings indicate that PEPT1 can transport midodrine and contributes to the high bioavailability of this drug and that Gly modification of DMAE is desirable for a prodrug of DMAE. Metabolism / Metabolites Thorough metabolic studies have not been conducted, but it appears that deglycination of midodrine to desglymidodrine takes place in many tissues, and both compounds are metabolized in part by the liver. The human cytochrome P450 (CYP) isoforms catalyzing the oxidation metabolism of desglymidodrine (DMAE), an active metabolite of midodrine, were studied. Recombinant human CYP2D6, 1A2 and 2C19 exhibited appreciable catalytic activity with respect to the 5'-O-demethylation of DMAE. The O-demethylase activity by the recombinant CYP2D6 was much higher than that of other CYP isoforms. Quinidine (a selective inhibitor of CYP2D6) inhibited the O-demethylation of DMAE in pooled human microsomes by 86%, while selective inhibitors for other forms of CYP did not show any appreciable effect. Although the activity of CYP2D6 was almost negligible in the PM microsomes, the O-demethylase activity of DMAE was found to be maintained by about 25% of the pooled microsomes. Furafylline (a selective inhibitor of CYP1A2) inhibited the M-2 formation in the PM microsomes by 57%. The treatment of pooled microsomes with an antibody against CYP2D6 inhibited the formation of M-2 by about 75%, whereas that of the PM microsomes did not show drastic inhibition. In contrast, the antibody against CYP1A2 suppressed the activity by 40 to 50% in the PM microsomes. These findings suggest that CYP2D6 have the highest catalytic activity of DMAE 5'-O-demethylation in human liver microsomes, followed by CYP1A2 to a small extent. Thorough metabolic studies have not been conducted, but it appears that deglycination of midodrine to desglymidodrine takes place in many tissues and both compounds are metabolized in part by the liver. Neither midodrine nor desglymidodrine is a substrate for monoamine oxidase. Biological Half-Life The metabolites display a half-life of about 3 to 4 hours. The plasma levels of the prodrug peak after about half an hour and decline with a half-life of approximately 25 minutes, while the metabolite reaches peak blood concentrations about 1 to 2 hours after a dose of midodrine and has a half-life of about 3 to 4 hours. |
|---|---|
| 毒性/毒理 (Toxicokinetics/TK) |
Interactions
It appears possible, although there is no supporting experimental evidence, that the high renal clearance of desglymidodrine (a base) is due to active tubular secretion by the base-secreting system also responsible for the secretion of such drugs as metformin, cimetidine, ranitidine, procainamide, triamterene, flecainide and quinidine. Thus there may be a potential for drug-drug interaction with these drugs. Midodrine hydrochloride has been used in patients concomitantly treated with salt-retaining steroid therapy (i.e., fludrocortisone acetate), with or without salt supplementation. The potential for supine hypertension should be carefully monitored in these patients and may be minimized by either reducing the dose of fludrocortisone acetate or decreasing the salt intake prior to initiation of treatment with midodrine hydrochloride. Alpha-adrenergic blocking agents, such as prazosin, terazosin and doxazosin, can antagonize the effects of midodrine hydrochloride. The use of drugs that stimulate alpha-adrenergic receptors (e.g., phenylephrine, pseudoephedrine, ephedrine, phenylpropanolamine or dihydroergotamine) may enhance or potentiate the pressor effects of midodrine hydrochloride. Therefore, caution should be used when midodrine hydrochloride is administered concomitantly with agents that cause vasoconstriction. When administered concomitantly with midodrine hydrochloride, cardiac glycosides may enhance or precipitate bradycardia, A.V. block or arrhythmia. An episode of transient, severe hypertension occurring within 2 minutes of injection of 1% lidocaine with 1:100,000 U of epinephrine in a patient taking midodrine for orthostatic hypotension /is reported/. /It was/ hypothesize that the patient's autonomic nervous system was dangerously susceptible to the effect of local anesthetic when combined with the vasoactive systemic effect of midodrine. Surgeons should minimize the use of vasoconstrictors in patients treated with midodrine to avoid hypertensive complications. Non-Human Toxicity Values LD50 Mouse oral 675 mg/kg LD50 Rat oral 30 to 50 mg/kg LD50 Dog oral 125-160 mg/kg |
| 其他信息 |
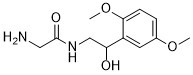
Midodrine is an aromatic ether that is 1,4-dimethoxybenzene which is substituted at position 2 by a 2-(glycylamino)-1-hydroxyethyl group. A direct-acting sympathomimetic with selective alpha-adrenergic agonist activity, it is used (generally as its hydrochloride salt) as a peripheral vasoconstrictor in the treatment of certain hypotensive states. The main active moiety is its major metabolite, deglymidodrine. It has a role as a prodrug, an alpha-adrenergic agonist, a sympathomimetic agent and a vasoconstrictor agent. It is a secondary alcohol, an amino acid amide and an aromatic ether. It is functionally related to a glycinamide and a deglymidodrine. It is a conjugate base of a midodrine(1+).
An ethanolamine derivative that is an adrenergic alpha agonist. It is used as a vasoconstrictor agent in the treatment of hypotension. Midodrine is an alpha-Adrenergic Agonist. The mechanism of action of midodrine is as an Adrenergic alpha-Agonist. Midodrine is a direct-acting prodrug and sympathomimetic agent with antihypotensive properties. Midodrine is converted to its active metabolite, desglymidodrine by deglycination reaction. Desglymidodrine selectively binds to and activates alpha-1-adrenergic receptors of the arteriolar and venous vasculature. This causes smooth muscle contraction and leads to an elevation of blood pressure. Desglymidodrine diffuses poorly across the blood-brain barrier, and is therefore not associated with effects on the central nervous system (CNS). An ethanolamine derivative that is an adrenergic alpha-1 agonist. It is used as a vasoconstrictor agent in the treatment of HYPOTENSION. See also: Midodrine Hydrochloride (has salt form). Drug Indication For the treatment of symptomatic orthostatic hypotension (OH). Mechanism of Action Midodrine undergoes metabolism to form its pharmacologically active metabolite, desglymidodrine. Desglymidodrine acts as an agonist at the alpha1-adrenergic receptors expressed in the arteriolar and venous vasculature. Activation of alpha1-adrenergic receptor signaling pathways lead to an increase in the vascular tone and elevation of blood pressure. Desglymidodrine is reported to have negligible effect on the cardiac beta-adrenergic receptors. Midodrine hydrochloride forms an active metabolite, desglymidodrine, that is an alpha1-agonist and exerts its actions via activation of the alpha-adrenergic receptors of the arteriolar and venous vasculature, producing an increase in vascular tone and elevation of blood pressure. Desglymidodrine does not stimulate cardiac beta-adrenergic receptors. Desglymidodrine diffuses poorly across the blood-brain barrier and is therefore not associated with effects on the central nervous system. Therapeutic Uses FDA proposed to withdraw approval of the drug midodrine hydrochloride, used to treat the low blood pressure condition, orthostatic hypotension, because required post-approval studies that verify the clinical benefit of the drug have not been done. To date, neither the original manufacturer nor any generic manufacturer has demonstrated the drug's clinical benefit, for example, by showing that use of the drug improved a patient's ability to perform life activities. ...used as a vasoconstrictor agent in the treatment of hypotension Midodrine hydrochloride is used in the management of symptomatic orthostatic hypotension; the drug is designated an orphan drug by the US Food and Drug Administration (FDA) for such use. /Included in US product label/ Drug Warnings WARNING: Because midodrine hydrochloride tablets can cause marked elevation of supine blood pressure, it should be used in patients whose lives are considerably impaired despite standard clinical care. The indication for use of midodrine hydrochloride tablets in the treatment of symptomatic orthostatic hypotension is based primarily on a change in a surrogate marker of effectiveness, an increase in systolic blood pressure measured one minute after standing, a surrogate marker considered likely to correspond to a clinical benefit. At present, however, clinical benefits of midodrine hydrochloride tablets, principally improved ability to carry out activities of daily living, have not been verified. The most potentially serious adverse reaction associated with midodrine hydrochloride therapy is marked elevation of supine arterial blood pressure (supine hypertension). Systolic pressure of about 200 mmHg was seen overall in about 13.4% of patients given 10 mg of midodrine hydrochloride. Systolic elevations of this degree were most likely to be observed in patients with relatively elevated pre-treatment systolic blood pressures (mean 170 mmHg). There is no experience in patients with initial supine systolic pressure above 180 mmHg, as those patients were excluded from the clinical trials. Use of midodrine hydrochloride in such patients is not recommended. Sitting blood pressures were also elevated by midodrine hydrochloride therapy. It is essential to monitor supine and sitting blood pressures in patients maintained on midodrine hydrochloride. The potential for supine and sitting hypertension should be evaluated at the beginning of midodrine hydrochloride therapy. Supine hypertension can often be controlled by preventing the patient from becoming fully supine, i.e., sleeping with the head of the bed elevated. The patient should be cautioned to report symptoms of supine hypertension immediately. Symptoms may include cardiac awareness, pounding in the ears, headache, blurred vision, etc. Midodrine hydrochloride use has not been studied in patients with hepatic impairment. Midodrine hydrochloride should be used with caution in patients with hepatic impairment, as the liver has a role in the metabolism of midodrine. For more Drug Warnings (Complete) data for Midodrine (12 total), please visit the HSDB record page. Pharmacodynamics Midodrine is a prodrug, i.e., the therapeutic effect of orally administered midodrine is due to the major metabolite desglymidodrine formed by deglycination of midodrine. Administration of midodrine results in a rise in standing, sitting, and supine systolic and diastolic blood pressure in patients with orthostatic hypotension of various etiologies. Standing systolic blood pressure is elevated by approximately 15 to 30 mmHg at 1 hour after a 10-mg dose of midodrine, with some effect persisting for 2 to 3 hours. Midodrine has no clinically significant effect on standing or supine pulse rates in patients with autonomic failure. |
| 分子式 |
C12H18N2O4
|
|---|---|
| 分子量 |
254.286
|
| 精确质量 |
254.127
|
| CAS号 |
42794-76-3
|
| 相关CAS号 |
Midodrine hydrochloride;43218-56-0;Midodrine-d6 hydrochloride;1188265-43-1
|
| PubChem CID |
4195
|
| 外观&性状 |
Typically exists as solid at room temperature
|
| LogP |
0.903
|
| tPSA |
93.81
|
| 氢键供体(HBD)数目 |
3
|
| 氢键受体(HBA)数目 |
5
|
| 可旋转键数目(RBC) |
6
|
| 重原子数目 |
18
|
| 分子复杂度/Complexity |
263
|
| 定义原子立体中心数目 |
0
|
| InChi Key |
MGCQZNBCJBRZDT-PPHPATTJSA-N
|
| InChi Code |
InChI=1S/C12H18N2O4.ClH/c1-17-8-3-4-11(18-2)9(5-8)10(15)7-14-12(16)6-13/h3-5,10,15H,6-7,13H2,1-2H3,(H,14,16)1H/t10-/m0./s1
|
| 化学名 |
(R)-2-amino-N-[2-(2,5-dimethoxyphenyl)-2-hydroxyethyl]acetamide hydrochloride
|
| 别名 |
St-1085 St1085 St 1085Amatine Pro-AmatineOrvaten
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples
|
|---|---|
| 溶解度 (体内实验) |
注意: 如下所列的是一些常用的体内动物实验溶解配方,主要用于溶解难溶或不溶于水的产品(水溶度<1 mg/mL)。 建议您先取少量样品进行尝试,如该配方可行,再根据实验需求增加样品量。
注射用配方
注射用配方1: DMSO : Tween 80: Saline = 10 : 5 : 85 (如: 100 μL DMSO → 50 μL Tween 80 → 850 μL Saline)(IP/IV/IM/SC等) *生理盐水/Saline的制备:将0.9g氯化钠/NaCl溶解在100 mL ddH ₂ O中,得到澄清溶液。 注射用配方 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (如: 100 μL DMSO → 400 μL PEG300 → 50 μL Tween 80 → 450 μL Saline) 注射用配方 3: DMSO : Corn oil = 10 : 90 (如: 100 μL DMSO → 900 μL Corn oil) 示例: 以注射用配方 3 (DMSO : Corn oil = 10 : 90) 为例说明, 如果要配制 1 mL 2.5 mg/mL的工作液, 您可以取 100 μL 25 mg/mL 澄清的 DMSO 储备液,加到 900 μL Corn oil/玉米油中, 混合均匀。 View More
注射用配方 4: DMSO : 20% SBE-β-CD in Saline = 10 : 90 [如:100 μL DMSO → 900 μL (20% SBE-β-CD in Saline)] 口服配方
口服配方 1: 悬浮于0.5% CMC Na (羧甲基纤维素钠) 口服配方 2: 悬浮于0.5% Carboxymethyl cellulose (羧甲基纤维素) 示例: 以口服配方 1 (悬浮于 0.5% CMC Na)为例说明, 如果要配制 100 mL 2.5 mg/mL 的工作液, 您可以先取0.5g CMC Na并将其溶解于100mL ddH2O中,得到0.5%CMC-Na澄清溶液;然后将250 mg待测化合物加到100 mL前述 0.5%CMC Na溶液中,得到悬浮液。 View More
口服配方 3: 溶解于 PEG400 (聚乙二醇400) 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 3.9325 mL | 19.6626 mL | 39.3252 mL | |
| 5 mM | 0.7865 mL | 3.9325 mL | 7.8650 mL | |
| 10 mM | 0.3933 mL | 1.9663 mL | 3.9325 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1


































