| 规格 | 价格 | |
|---|---|---|
| 500mg | ||
| 1g | ||
| Other Sizes |

| 靶点 |
β1 adrenoceptor
|
|---|---|
| 体外研究 (In Vitro) |
美托洛尔(0-1000 μg/mL;24-72 小时)对 MOLT-4 和 U937 细胞的细胞毒性作用具有剂量和时间依赖性 [3]。
|
| 体内研究 (In Vivo) |
在 ApoE−/− 小鼠中,美托洛尔(2.5 mg/kg/h;输注;11 周)可减少动脉粥样硬化和促炎细胞因子 [1]。美托洛尔(15 mg/kg/q12h;ig;5 天)在由柯萨奇病毒 B3 引起的病毒性心肌炎小鼠模型中显示出抗病毒和抗炎特性 [2]。在患有冠状动脉微栓塞(CME)的大鼠中,美托洛尔(2.5 mg/kg;静脉注射;3次推注)有效防止心肌细胞死亡并减少活化的caspase-9蛋白表达[4]。
|
| 细胞实验 |
细胞毒性测定 [3]
细胞类型: U937 和 MOLT-4 细胞 测试浓度: 1、10、50、100、500 和 1000 μg/ mL 孵育持续时间:24、48 和 72 小时 实验结果:孵育的 U937 和 MOLT -4 细胞的活力显着降低在 1000 μg/mL (3740.14μM) 浓度下孵育 48 小时 (hrs (hours)) 在 ≥500 μg/ml (≥1870.07μM) 浓度下孵育 72 小时 (hrs (hrs) 后,U937 细胞的活力显着降低小时)),并且在孵育 72 小时后,U937 细胞的活力显着降低。 hrs(小时)后,MOLT4细胞浓度≥100 μg/ml(≥374.01μM)。 |
| 动物实验 |
Animal/Disease Models: Male ApoE−/− mice [1]
Doses: 2.5 mg/kg/h Route of Administration: via mini-osmotic pump, 11 weeks Experimental Results: Thoracic aorta atherosclerotic plaque area Dramatically diminished, serum TNFα and chemokine CXCL1, and diminished macrophage content in plaques. Animal/Disease Models: Balb/c mouse, coxsackie virus B3 (CVB3)-induced viral myocarditis (VMC) model [2] Doses: 15 mg/kg/q12h Route of Administration: po (oral gavage), for 5 days Experimental Results: CVB3 infection-induced reduction in VMC pathology score protects myocardium from viral damage by reducing serum cTn-I levels. Reduce myocardial pro-inflammatory cytokine levels and increase anti-inflammatory cytokine expression. Myocardial virus titers were Dramatically diminished. |
| 药代性质 (ADME/PK) |
Absorption
When metoprolol is administered orally, it is almost completely absorbed in the gastrointestinal tract. The maximum serum concentration is achieved 20 min after intravenous administration and 1-2 hours after oral administration. The bioavailability of metoprolol is of 100% when administered intravenously and when administered orally it presents about 50% for the tartrate derivative and 40% for the succinate derivative. The absorption of metoprolol in the form of the tartrate derivative is increased by the concomitant administration of food. Route of Elimination Metoprolol is mainly excreted via the kidneys. From the eliminated dose, less than 5% is recovered unchanged. Volume of Distribution The reported volume of distribution of metoprolol is 4.2 L/kg. Due to the characteristics of metoprolol, this molecule is able to cross the blood-brain barrier and even 78% of the administered drug can be found in cerebrospinal fluid. Clearance The reported clearance rate on patients with normal kidney function is 0.8 L/min. In cirrhotic patients, the clearance rate changes to 0.61 L/min. Plasma levels following oral administration of conventional metoprolol tablets, however, approximate 50% of levels following intravenous adminsitration, indicating about 50% first-pass metabolism... Elimination is mainly by biotransformation in the liver. View More
Metoprolol tartrate is rapidly and almost completely absorbed from the GI tract; absorption of a single oral dose of 20-100 mg is complete in 2.5-3 hours. After an oral dose, about 50% of the drug administered as conventional tablets appears to undergo first-pass metabolism in the liver. Bioavailability of orally administered metoprolol tartrate increases with increased doses, indicating a possible saturable disposition process of low capacity such as tissue binding in the liver. Steady-state oral bioavailability of extended-release tablets of metoprolol succinate given once daily at dosages equivalent to 50-400 mg of metoprolol tartrate is about 77% of that of conventional tablets at corresponding dosages given once daily or in divided doses. Food does not appear to affect bioavailability of metoprolol succinate extended-release tablets. Following a single oral dose as conventional tablets, metoprolol appears in the plasma within 10 minutes and peak plasma concentrations are reached in about 90 minutes. When metoprolol tartrate conventional tablets are administered with food rather than on an empty stomach, peak plasma concentrations are higher and the extent of absorption of the drug is increased. Following oral administration of metoprolol succinate as extended-release tablets, peak plasma metoprolol concentrations are aobut 25-50% of those attained after administration of metoprolol tartrate conventional tablets given once daily or in divided doses. Time to peak concentration is longer with extended-release tablets, with peak plasma coentrations being reached in about 7 hours following administration of such tablets. Plasma concentrations attained 1 hour after an oral dose are linearly related to metoprolol tartrate doses ranging from 50-400 mg as conventional tablets.
Metabolism / Metabolites Metoprolol goes through significant first-pass hepatic metabolism which covers around 50% of the administered dose. The metabolism of metoprolol is mainly driven by the activity of CYP2D6 and to a lesser extent due to the activity of CYP3A4. The metabolism of metoprolol is mainly represented by reactions of hydroxylation and O-demethylation. Metoprolol does not inhibit or enhance its own metabolism. Three main metabolites of the drug are formed by oxidative deamination, O-dealkylation with subsequent oxidation, and aliphatic hydroxylation; these metabolites account for 85% of the total urinary excretion of metabolites. The metabolites apparently do not have appreciable pharmacologic activity. The rate of hydroxylation, resulting in alpha-hydroxymetoprolol, is genetically determined and is subject to considerable interindividual variation. Poor hydroxylators of metoprolol have increased areas under the plasma concentration-time curves, prolonged elimination half-lives (about 7.6 hours), higher urinary concentrations of unchanged drug, and negligible urinary concentrations of alpha-hydroxymetoprolol compared with extensive hydroxylators. Beta-adrenergic blockade of exercise-induced tachycardia persists for at least 24 hours after administration of a single 200-mg oral dose of metoprolol tartrate in poor hydroxylators. Controlled studies have shown that debrisoquine oxidation phenotype is a major determinant of the metabolism, pharmacokinetics and some of the pharmacological actions of metoprolol. The poor metabolizer phenotype is associated with increased plasma drug concentrations, a prolongation of elimination half-life and more intense and sustained beta blockade. Phenotypic differences have also been observed in the pharmacokinetics of the enantiomers of metoprolol. In vivo and in vitro studies have identified some of the metabolic pathways which are subject to the defect, that is alpha-hydroxylation and O-demethylation. PMID:2868819 Metropolol is a racemic mixture of R-and S-enantiomers, and is primarily metabolized by CYP2D6. Biological Half-Life The immediate release formulations of metoprolol present a half-life of about 3-7 hours. The plasma half-life ranges from approximately 3 to 7 hours. |
| 毒性/毒理 (Toxicokinetics/TK) |
Hepatotoxicity
Metoprolol therapy has been associated with a low rate of mild-to-moderate elevations of serum aminotransferase levels which are usually asymptomatic and transient and resolve even with continuation of therapy. A few instances of clinically apparent, acute liver injury attributable to metoprolol have been reported. In view of its wide scale use, metoprolol induced liver injury is exceedingly rare. The typical liver injury associated with beta-blockers has a latency to onset of 2 to 12 weeks and a hepatocellular pattern of liver enzyme. Symptoms of hypersensitivity (rash, fever, eosinophilia) and autoantibody formation have not been reported. Reported cases due to metoprolol have included cases of acute liver failure, but ultimately all were self-limiting and resolved fairly rapidly once once drug was stopped. Likelihood score: D (possible rare cause of clinically apparent liver injury). Effects During Pregnancy and Lactation ◉ Summary of Use during Lactation Because of the low levels of metoprolol in breastmilk, amounts ingested by the infant are small and would not be expected to cause any adverse effects in breastfed infants. Studies on the use of metoprolol during breastfeeding have found no adverse reactions in breastfed infants. Monitor breastfed infants for symptoms of beta blockade such as bradycardia and listlessness due to hypoglycemia. ◉ Effects in Breastfed Infants A study of mothers taking beta-blockers during nursing found a numerically, but not statistically significant increased number of adverse reactions in those taking any beta-blocker. Although the ages of infants were matched to control infants, the ages of the affected infants were not stated. Of 6 mothers taking metoprolol, none reported adverse effects in her breastfed infant. ◉ Effects on Lactation and Breastmilk Relevant published information on the effects of beta-blockade or metoprolol during normal lactation was not found as of the revision date. A study in 6 patients with hyperprolactinemia and galactorrhea found no changes in serum prolactin levels following beta-adrenergic blockade with propranolol. View More
◈ What is metoprolol?
Interactions The effect of verapamil coadministration on the hepatic first pass clearance of metoprolol was investigated in dogs. Plasma concentration-time course of metoprolol enantiomers and urinary recovery of oxidative metabolites were determined after a single iv (0.51 mg/kg) and an oral (1.37 mg/kg) dose of deuterium labeled pseudoracemic metoprolol, with or without concomitant administration of racemic verapamil (3 mg/kg). Verapamil inhibited both the systemic and oral clearance of metoprolol by about 50-70%. The first pass effect of metoprolol was completely abolished after coadministration of verapamil, reflecting a marked alteration in the degree of hepatic extraction of metoprolol from intermediate to low. The hepatic clearance of metoprolol was slightly (S)-enantioselective (R/S ratio = 0.89 + or - 0.04) in control dogs. Inhibition of hepatic clearance of metoprolol by verapamil was selective towards (S)-metoprolol, such that the enantioselectivity in hepatic clearance toward (S)-metoprolol disappeared following verapamil coadministration (R/S ratio = 1.01 + or - 0.05). Urinary metabolite profiles indicated that O-demethylation and N-dealkylation were the major pathways of oxidative metabolism in the dog. alpha-Hydroxymetoprolol was a minor metabolite in urine. N-Dealkylation showed a strong preference for (S)-metoprolol, whereas O-demethylation and alpha-hydroxylation exhibited a modest selectivity toward (R)-metoprolol; hence, the slight (S)-enantioselectivity in the overall hepatic clearance. Comparison of metoprolol metabolite formation clearances in the absence or presence of verapamil coadministration showed that all three oxidative pathways were inhibited by 60-80%. The greater inhibition of hepatic clearance observed with (S)-metoprolol as compared to (R)-metoprolol was attributed to a significant (S)-enantioselective inhibition in the O-demethylation of metoprolol by verapamil. PMID:1687016 The interaction between metoprolol and bromazepam and lorazepam was studied in 12 healthy male volunteers aged 21-37 years. Metoprolol had no significant effect on the pharmacokinetics of bromazepam or lorazepam. However, bromazepam area under the curve was 35% higher in the presence of metoprolol. Bromazepam enhanced the effect of metoprolol on systolic blood pressure but not on diastolic blood pressure or pulse rate. Lorazepam had no effect on either blood pressure or pulse. Metoprolol did not enhance the effect of bromazepam on the psychomotor tests used in this study. Metoprolol caused a small increase in critical flicker fusion threshold with lorazepam but had no effect on the other tests. Lorazepam (2 mg) was more potent than bromazepam (6 mg) in the doses used in this study. The interaction of metoprolol with bromazepam and lorazepam is unlikely to be of clinical significance. No change in dose is necessary when using these drugs together. Protein Binding Metoprolol is not highly bound to plasma proteins and only about 11% of the administered dose is found bound. It is mainly bound to serum albumin. |
| 参考文献 | |
| 其他信息 |
Metoprolol Fumarate is the fumarate salt form of metoprolol, a cardioselective competitive beta-1 adrenergic receptor antagonist with antihypertensive properties and devoid of intrinsic sympathomimetic activity. Metoprolol antagonizes beta 1-adrenergic receptors in the myocardium, thereby reducing the rate and force of myocardial contraction, and consequently a diminished cardiac output. This agent may also reduce the secretion of renin with subsequent reduction in levels of angiotensin II thereby preventing vasoconstriction and aldosterone secretion.
See also: Metoprolol (has active moiety). |
| 分子式 |
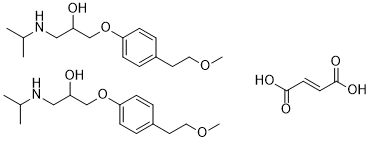
2[C15H25NO3].C4H4O4
|
|---|---|
| 分子量 |
650.79996
|
| 精确质量 |
383.194
|
| 元素分析 |
C, 62.75; H, 8.36; N, 4.30; O, 24.58
|
| CAS号 |
80274-67-5
|
| 相关CAS号 |
Metoprolol succinate;98418-47-4;Metoprolol-d7 hydrochloride;1219798-61-4; Metoprolol tartrate;56392-17-7;Metoprolol-d7;959787-96-3;(R)-Metoprolol-d7;1292907-84-6;(S)-Metoprolol-d7;1292906-91-2;Metoprolol-d5;959786-79-9; 51384-51-1; 56392-18-8 (HCl); 80274-67-5 (fumarate)
|
| PubChem CID |
6440651
|
| 外观&性状 |
Typically exists as solid at room temperature
|
| tPSA |
176
|
| 氢键供体(HBD)数目 |
6
|
| 氢键受体(HBA)数目 |
12
|
| 可旋转键数目(RBC) |
20
|
| 重原子数目 |
46
|
| 分子复杂度/Complexity |
334
|
| 定义原子立体中心数目 |
0
|
| SMILES |
COCCC1C=CC(OCC(CNC(C)C)O)=CC=1.COCCC1C=CC(OCC(CNC(C)C)O)=CC=1.OC(=O)/C=C/C(=O)O
|
| InChi Key |
BRIPGNJWPCKDQZ-WXXKFALUSA-N
|
| InChi Code |
InChI=1S/2C15H25NO3.C4H4O4/c2*1-12(2)16-10-14(17)11-19-15-6-4-13(5-7-15)8-9-18-3;5-3(6)1-2-4(7)8/h2*4-7,12,14,16-17H,8-11H2,1-3H3;1-2H,(H,5,6)(H,7,8)/b;;2-1+
|
| 化学名 |
(E)-but-2-enedioic acid;1-[4-(2-methoxyethyl)phenoxy]-3-(propan-2-ylamino)propan-2-ol
|
| 别名 |
Metoprolol fumarate; 80274-67-5; Lopresor OROS; CGP 2175C; Lopressor ORO; UNII-IO1C09Z674; 119637-66-0; LOPRESSOR OROS;
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples
|
|---|---|
| 溶解度 (体内实验) |
注意: 如下所列的是一些常用的体内动物实验溶解配方,主要用于溶解难溶或不溶于水的产品(水溶度<1 mg/mL)。 建议您先取少量样品进行尝试,如该配方可行,再根据实验需求增加样品量。
注射用配方
注射用配方1: DMSO : Tween 80: Saline = 10 : 5 : 85 (如: 100 μL DMSO → 50 μL Tween 80 → 850 μL Saline)(IP/IV/IM/SC等) *生理盐水/Saline的制备:将0.9g氯化钠/NaCl溶解在100 mL ddH ₂ O中,得到澄清溶液。 注射用配方 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (如: 100 μL DMSO → 400 μL PEG300 → 50 μL Tween 80 → 450 μL Saline) 注射用配方 3: DMSO : Corn oil = 10 : 90 (如: 100 μL DMSO → 900 μL Corn oil) 示例: 以注射用配方 3 (DMSO : Corn oil = 10 : 90) 为例说明, 如果要配制 1 mL 2.5 mg/mL的工作液, 您可以取 100 μL 25 mg/mL 澄清的 DMSO 储备液,加到 900 μL Corn oil/玉米油中, 混合均匀。 View More
注射用配方 4: DMSO : 20% SBE-β-CD in Saline = 10 : 90 [如:100 μL DMSO → 900 μL (20% SBE-β-CD in Saline)] 口服配方
口服配方 1: 悬浮于0.5% CMC Na (羧甲基纤维素钠) 口服配方 2: 悬浮于0.5% Carboxymethyl cellulose (羧甲基纤维素) 示例: 以口服配方 1 (悬浮于 0.5% CMC Na)为例说明, 如果要配制 100 mL 2.5 mg/mL 的工作液, 您可以先取0.5g CMC Na并将其溶解于100mL ddH2O中,得到0.5%CMC-Na澄清溶液;然后将250 mg待测化合物加到100 mL前述 0.5%CMC Na溶液中,得到悬浮液。 View More
口服配方 3: 溶解于 PEG400 (聚乙二醇400) 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.5366 mL | 7.6829 mL | 15.3657 mL | |
| 5 mM | 0.3073 mL | 1.5366 mL | 3.0731 mL | |
| 10 mM | 0.1537 mL | 0.7683 mL | 1.5366 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
| NCT Number | Recruitment | interventions | Conditions | Sponsor/Collaborators | Start Date | Phases |
| NCT02123056 | Active Recruiting |
Drug: Metoprolol Drug: Matching Placebo |
Vasovagal Syncope | University of Calgary | October 2014 | Phase 4 |
| NCT01608893 | Active Recruiting |
Drug: Carvedilol Drug: Metoprolol |
Atrial Fibrillation | University of Calgary | May 2012 | Not Applicable |
| NCT03278509 | Active Recruiting |
Drug: Metoprolol Succinate Drug: Bisoprolol |
Acute Myocardial InfarctionST Elevation Myocardial Infarction |
Karolinska Institutet | September 11, 2017 | Phase 4 |
| NCT03070184 | Active Recruiting |
Other: Exercise challenge Drug: Metoprolol Succinate ER |
Healthy Pre Hypertension |
University of Alabama at Birmingham |
April 30, 2017 | Phase 2 |
| NCT05741385 | Recruiting | Drug: Caffeine Drug: Warfarin sodium Drug: Omeprazole Drug: Metoprolol |
Liver Cirrhosis | Boehringer Ingelheim | April 25, 2023 | Not Applicable |
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1

































