
| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 100mg |
|
||
| 250mg |
|
||
| 500mg |
|
||
| 1g |
|
||
| 2g |
|
||
| 5g |
|
||
| Other Sizes |
|

| 靶点 |
5-HT3 Receptor ( IC50 = 308 nM ); D2 Receptor ( IC50 = 483 nM )
|
|---|---|
| 体外研究 (In Vitro) |
体外活性:甲氧氯普胺是一种多巴胺受体拮抗剂,在过去三十年中已用于治疗各种胃肠道症状。在各国,甲氧氯普胺是孕妇首选的止吐药物。研究结果让女性在怀孕期间服用甲氧氯普胺缓解恶心和呕吐时对胎儿的安全性感到放心。有证据还支持其用于治疗胃轻瘫(胃排空不良)和胃食管反流病。它似乎与多巴胺 D2 受体结合,是一种受体拮抗剂,也是一种混合的 5-HT3 受体拮抗剂/5-HT4 受体激动剂。
|
| 体内研究 (In Vivo) |
甲氧氯普胺(6.7 µg/g;每天一次,皮下注射,持续 50 天)在动情周期的每个阶段,盐酸都会显着提高垂体催乳素细胞的数量和体积[4]。
甲氧氯普胺盐酸盐(5–40 mg/kg) ;腹膜内)导致僵直和拮抗 小鼠攀爬笼子的倾向是由阿朴吗啡诱导的[5]。 盐酸甲氧氯普胺(1.25-2.5 mg/kg;腹膜内注射)会诱导小鼠出现攀爬笼子的刻板行为[5]。 |
| 动物实验 |
Adult, virgin female mice of the Swiss EPM-1 strain
6.7 µg/g S.c. daily for 50 days |
| 药代性质 (ADME/PK) |
Absorption, Distribution and Excretion
Metoclopramide is rapidly absorbed in the gastrointestinal tract with an absorption rate of about 84%. The bioavailability of the oral preparation is reported to be about 40.7%, but can range from 30-100%. Nasal metoclopramide is 47% bioavailable. A 15mg dose reaches a Cmax of 41.0 ng/mL, with a Tmax of 1.25 h, and an AUC of 367 ng\*h/mL. About 85% of an orally administered dose was measured in the urine within 72 hours during a pharmacokinetic study. An average of 18% to 22% of 10-20 mg dose was recovered as free drug within 3 days of administration. The volume of distribution of metoclopramide is approximately 3.5 L/kg. This implies a high level of tissue distribution. Metoclopramide crosses the placental barrier and can cause extrapyramidal symptoms in the fetus. The renal clearance of metoclopramide is 0.16 L/h/kg with a total clearance of 0.7 L/h/kg. Clinical studies showed that the clearance of metoclopramide may be reduced by up to 50% in patients with renal impairment. After high intravenous doses, total metoclopramide clearance ranged from 0.31 to 0.69 L/kg/h. Metoclopramide is rapidly and almost completely absorbed from the GI tract following oral administration; however, absorption may be delayed or diminished in patients with gastric stasis. Considerable interindividual variations (up to fivefold) in peak plasma concentration have been reported with the same oral dose of metoclopramide. This variability apparently results from interindividual differences in first-pass metabolism of the drug. Bioavailability of metoclopramide appears to correlate with the ratio of free:conjugated metoclopramide concentrations in urine. It appears that sulfate conjugation in the GI lumen and/or during first pass through the liver is the principal determinant of bioavailability of orally administered metoclopramide. The absolute bioavailability of orally administered metoclopramide has not been clearly established in humans, but limited data indicate that 30-100% of an oral dose of the drug reaches systemic circulation as unchanged metoclopramide. Following IM administration, the absolute bioavailability of metoclopramide is 74-96%. Following oral administration of a single 10-mg dose of the drug in healthy, fasting adults in one study, peak plasma metoclopramide concentrations of 32-44 ng/mL occurred at 1-2 hours; following oral administration of a single 20-mg dose, peak plasma metoclopramide concentrations of 72-87 ng/mL occurred at an average of 2 hours. In a study in infants (3.5 weeks-5.4 months of age) with gastroesophageal reflux who received 0.15-mg/kg oral doses of metoclopramide every 6 hours for 10 doses as an oral solution, the mean peak plasma concentration (56.8 ng/mL) of the drug after the 10th dose was twofold higher compared with that after the first dose (29 ng/mL), suggesting that metoclopramide accumulates in plasma following multiple oral dosing in this age group. In these patients, time to reach mean peak plasma concentrations (2.2 hours) was similar after the 10th dose to that occurring after the first dose. For more Absorption, Distribution and Excretion (Complete) data for Metoclopramide (18 total), please visit the HSDB record page. Metabolism / Metabolites Metoclopramide undergoes first-pass metabolism and its metabolism varies according to the individual. This drug is metabolized by cytochrome P450 enzymes in the liver. CYP2D6 and CYP3A4 both contribute to its metabolism, with CYP2D6 being more heavily involved. CYP1A2 is also a minor contributing enzyme. The process of N-4 sulphate conjugation is a primary metabolic pathway of metoclopramide. Although the exact metabolic fate of metoclopramide is not clearly established, it appears that metoclopramide is only minimally metabolized. The major metabolite found in urine is 2-[(4-amino-5-chloro-2-methoxybenzoyl)amino]acetic acid; it is not known if this metabolite is pharmacologically active. Metoclopramide is conjugated with sulfuric and/or glucuronic acid. Metoclopramide has known human metabolites that include monodeethylmetoclopramide. Biological Half-Life The mean elimination half-life of metoclopramide in people with healthy renal function ranges from 5 to 6 hours but is prolonged in patients with renal impairment. Downward dose adjustment should be considered. In adults, the half-life of metoclopramide in the initial phase (t1/2 alpha) is about 5 minutes, and the half-life in the terminal phase (t1/2 beta) ranges from 2.5-6 hours. In children receiving oral or IV metoclopramide, the elimination half-life of the drug reportedly is 4.1-4.5 hours. Following oral administration of 0.15-mg/kg doses of metoclopramide every 6 hours for 10 doses in an infant (3.5 weeks of age), elimination half-lives of 23.1 and 10.3 hours were observed after the first and 10th dose, respectively, which were substantially longer than those reported in older infants, suggesting a reduced clearance in the neonate possibly being associated with immature renal and hepatic functions present at birth. |
| 毒性/毒理 (Toxicokinetics/TK) |
Interactions
The effects of metoclopramide on gastrointestinal motility are antagonized by anticholinergic drugs and narcotic analgesics. Additive sedative effects can occur when metoclopramide is given with alcohol, sedatives, hypnotics, narcotics, or tranquilizers. The finding that metoclopramide releases catecholamines in patients with essential hypertension suggests that it should be used cautiously, if at all, in patients receiving monoamine oxidase inhibitors. Absorption of drugs from the stomach may be diminished (e.g., digoxin) by metoclopramide, whereas the rate and/or extent of absorption of drugs from the small bowel may be increased (e.g., acetaminophen, tetracycline, levodopa, ethanol, cyclosporine). Gastroparesis (gastric stasis) may be responsible for poor diabetic control in some patients. Exogenously administered insulin may begin to act before food has left the stomach and lead to hypoglycemia. Because the action of metoclopramide will influence the delivery of food to the intestines and thus the rate of absorption, insulin dosage or timing of dosage may require adjustment. For more Interactions (Complete) data for Metoclopramide (10 total), please visit the HSDB record page. Non-Human Toxicity Values LD50 Rat oral 750 mg/kg LD50 Rat ip 114 mg/kg LD50 Rat sc 340 mg/kg LD50 Rat iv 50 mg/kg For more Non-Human Toxicity Values (Complete) data for Metoclopramide (8 total), please visit the HSDB record page. |
| 参考文献 |
|
| 其他信息 |
Therapeutic Uses
Antiemetics; Dopamine Antagonists Metoclopramide tablets are indicated as short-term (4 to 12 weeks) therapy for adults with symptomatic, documented gastroesophageal reflux who fail to respond to conventional therapy. /Included in US product label/ Metoclopramide tablets, USP is indicated for the relief of symptoms associated with acute and recurrent diabetic gastric stasis. The usual manifestations of delayed gastric emptying (eg, nausea, vomiting, heartburn, persistent fullness after meals, and anorexia) appear to respond to Metoclopramide Tablets within different time intervals. Significant relief of nausea occurs early and continues to improve over a three-week period. Relief of vomiting and anorexia may precede the relief of abdominal fullness by one week or more. /Included in US product label/ Metoclopramide injection is indicated for the prophylaxis of vomiting associated with emetogenic cancer chemotherapy. /Included in US product label/ For more Therapeutic Uses (Complete) data for Metoclopramide (8 total), please visit the HSDB record page. Drug Warnings WARNING: TARDIVE DYSKINESIA-Treatment with metoclopramide can cause tardive dyskinesia, a serious movement disorder that is often irreversible. The risk of developing tardive dyskinesia increases with duration of treatment and total cumulative dose. Metoclopramide therapy should be discontinued in patients who develop signs or symptoms of tardive dyskinesia. There is no known treatment for tardive dyskinesia. In some patients, symptoms may lessen or resolve after metoclopramide treatment is stopped. Treatment with metoclopramide for longer than 12 weeks should be avoided in all but rare cases where therapeutic benefit is thought to outweigh the risk of developing tardive dyskinesia. Adverse reactions to metoclopramide generally involve the CNS and GI tract and are usually mild, transient, and reversible following discontinuance of the drug. In general, the incidence of metoclopramide-induced adverse effects is related to dosage and duration of therapy. The most frequent adverse effects of metoclopramide involve the CNS. Restlessness, drowsiness, fatigue, and lassitude have been reported in patients receiving the drug; these effects occur in about 10% of patients receiving a dosage of 10 mg 4 times daily. Insomnia, headache, confusion, dizziness, or depression with suicidal ideation occurs less frequently. The risk of drowsiness is increased at higher doses, occurring in about 70% of patients receiving doses of 1-2 mg/kg. Seizures have been reported rarely, although a causal relationship to metoclopramide has not been established. Hallucinations also have been reported rarely. Feelings of anxiety or agitation also may occur, especially following rapid IV injection of the drug. Extrapyramidal reactions (eg, acute dystonic reactions, akathisia) may occur in patients receiving metoclopramide and apparently are mediated via blockade of central dopaminergic receptors involved in motor function. Although extrapyramidal reactions may occur in all age groups and at any dose, they occur more frequently in pediatric patients and adults younger than 30 years of age and following IV administration of high doses of the drug (eg, those used in prophylaxis of cancer chemotherapy-induced vomiting). Extrapyramidal reactions generally occur within 24-48 hours after starting therapy and usually subside within 24 hours following discontinuance of the drug. For more Drug Warnings (Complete) data for Metoclopramide (31 total), please visit the HSDB record page. Pharmacodynamics Metoclopramide increases gastric emptying by decreasing lower esophageal sphincter (LES) pressure. It also exerts effects on the area postrema of the brain, preventing and relieving the symptoms of nausea and vomiting. In addition, this drug increases gastrointestinal motility without increasing biliary, gastric, or pancreatic secretions. Because of its antidopaminergic activity, metoclopramide can cause symptoms of tardive dyskinesia (TD), dystonia, and akathisia, and should therefore not be administered for longer than 12 weeks. |
| 分子式 |
C14H23CL2N3O2
|
|
|---|---|---|
| 分子量 |
336.26
|
|
| 精确质量 |
335.117
|
|
| 元素分析 |
C, 50.01; H, 6.89; Cl, 21.09; N, 12.50; O, 9.52
|
|
| CAS号 |
7232-21-5
|
|
| 相关CAS号 |
Metoclopramide; 364-62-5; Metoclopramide hydrochloride hydrate; 54143-57-6
|
|
| PubChem CID |
4168
|
|
| 外观&性状 |
Solid powder
|
|
| 沸点 |
418.7ºC at 760 mmHg
|
|
| 熔点 |
145ºC
|
|
| 闪点 |
207ºC
|
|
| LogP |
3.776
|
|
| tPSA |
67.59
|
|
| 氢键供体(HBD)数目 |
2
|
|
| 氢键受体(HBA)数目 |
4
|
|
| 可旋转键数目(RBC) |
7
|
|
| 重原子数目 |
20
|
|
| 分子复杂度/Complexity |
300
|
|
| 定义原子立体中心数目 |
0
|
|
| SMILES |
CCN(CCNC(C1=CC(Cl)=C(N)C=C1OC)=O)CC.Cl
|
|
| InChi Key |
RVFUNJWWXKCWNS-UHFFFAOYSA-N
|
|
| InChi Code |
InChI=1S/C14H22ClN3O2.ClH/c1-4-18(5-2)7-6-17-14(19)10-8-11(15)12(16)9-13(10)20-3;/h8-9H,4-7,16H2,1-3H3,(H,17,19);1H
|
|
| 化学名 |
4-amino-5-chloro-N-[2-(diethylamino)ethyl]-2-methoxybenzamide;hydrochloride
|
|
| 别名 |
|
|
| HS Tariff Code |
2934.99.9001
|
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
|
|||
|---|---|---|---|---|
| 溶解度 (体内实验) |
注意: 如下所列的是一些常用的体内动物实验溶解配方,主要用于溶解难溶或不溶于水的产品(水溶度<1 mg/mL)。 建议您先取少量样品进行尝试,如该配方可行,再根据实验需求增加样品量。
注射用配方
注射用配方1: DMSO : Tween 80: Saline = 10 : 5 : 85 (如: 100 μL DMSO → 50 μL Tween 80 → 850 μL Saline)(IP/IV/IM/SC等) *生理盐水/Saline的制备:将0.9g氯化钠/NaCl溶解在100 mL ddH ₂ O中,得到澄清溶液。 注射用配方 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (如: 100 μL DMSO → 400 μL PEG300 → 50 μL Tween 80 → 450 μL Saline) 注射用配方 3: DMSO : Corn oil = 10 : 90 (如: 100 μL DMSO → 900 μL Corn oil) 示例: 以注射用配方 3 (DMSO : Corn oil = 10 : 90) 为例说明, 如果要配制 1 mL 2.5 mg/mL的工作液, 您可以取 100 μL 25 mg/mL 澄清的 DMSO 储备液,加到 900 μL Corn oil/玉米油中, 混合均匀。 View More
注射用配方 4: DMSO : 20% SBE-β-CD in Saline = 10 : 90 [如:100 μL DMSO → 900 μL (20% SBE-β-CD in Saline)] 口服配方
口服配方 1: 悬浮于0.5% CMC Na (羧甲基纤维素钠) 口服配方 2: 悬浮于0.5% Carboxymethyl cellulose (羧甲基纤维素) 示例: 以口服配方 1 (悬浮于 0.5% CMC Na)为例说明, 如果要配制 100 mL 2.5 mg/mL 的工作液, 您可以先取0.5g CMC Na并将其溶解于100mL ddH2O中,得到0.5%CMC-Na澄清溶液;然后将250 mg待测化合物加到100 mL前述 0.5%CMC Na溶液中,得到悬浮液。 View More
口服配方 3: 溶解于 PEG400 (聚乙二醇400) 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.9739 mL | 14.8694 mL | 29.7389 mL | |
| 5 mM | 0.5948 mL | 2.9739 mL | 5.9478 mL | |
| 10 mM | 0.2974 mL | 1.4869 mL | 2.9739 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
The Effect of Dopamine on Pulmonary Diffusion and Capillary Blood Volume During Exercise
CTID: NCT02965963
Phase: N/A Status: Active, not recruiting
Date: 2024-09-19
|
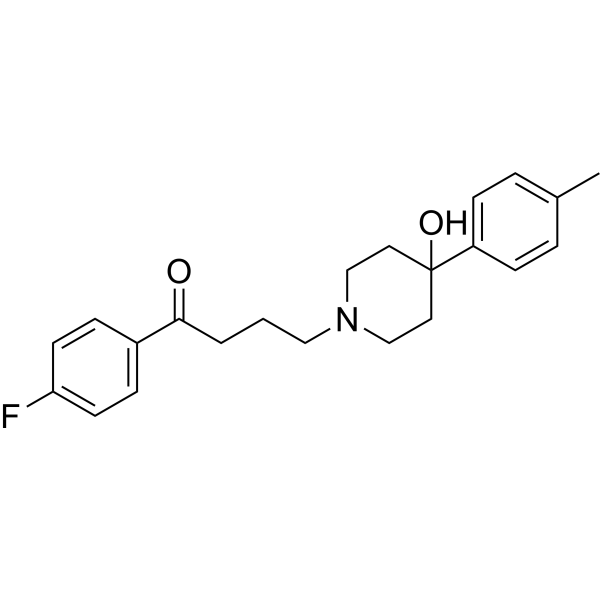 Moperone
Moperone
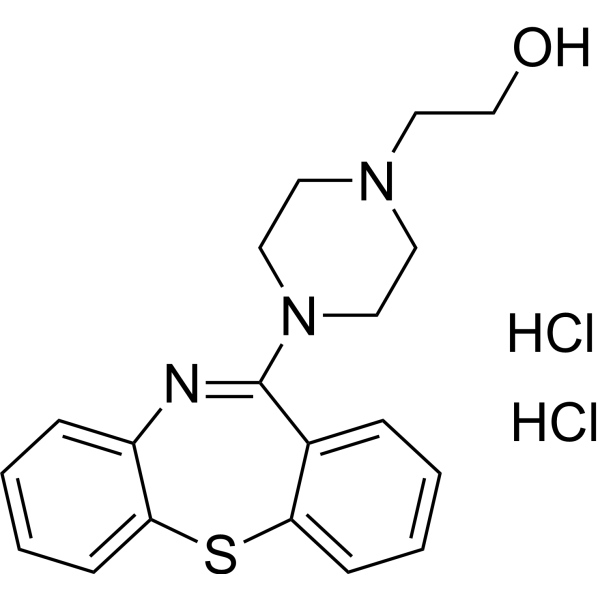 Desethoxy Quetiapine dihydrochloride
Desethoxy Quetiapine dihydrochloride
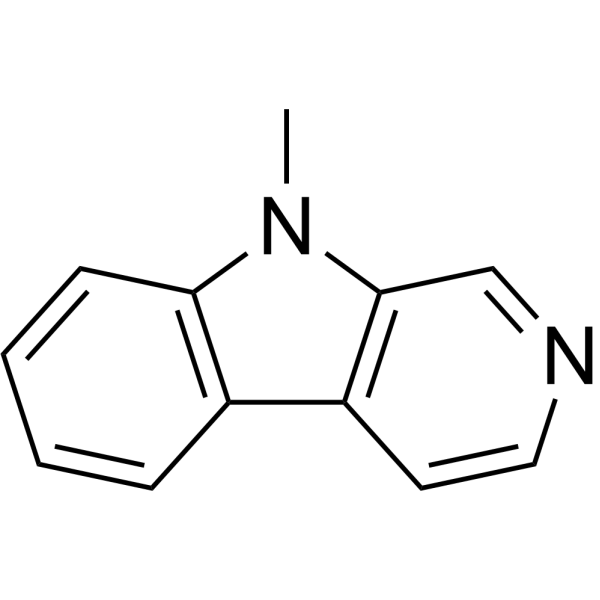 9-Methyl-β-carboline
9-Methyl-β-carboline
 S-Secoantioquine
S-Secoantioquine
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1





























 COA
COA

