| 规格 | 价格 | |
|---|---|---|
| 500mg | ||
| 1g | ||
| Other Sizes |

| 体外研究 (In Vitro) |
Melflufen(美法仑氟芬酰胺)以浓度依赖性方式降低 MM.1S、INA-6、RPMI-8226、MM.1R、Dox-40、ARP-1 和 ANBL-6 细胞的活力 (0.5-10 μM;24 小时)[1]。在 MM.1S 细胞中,melflufen 会导致细胞凋亡 [1]。此外,马法兰是一种强效的外泌体分泌激活剂[3]。
|
|---|---|
| 体内研究 (In Vivo) |
在异种移植小鼠模型中,melflufen(3 mg/kg;每周静脉注射两次,持续两周)表现出抗 MM 作用 [1]。
|
| 细胞实验 |
细胞活力测定
细胞类型: 多发性骨髓瘤细胞:MM.1S、INA-6、RPMI-8226、MM.1R、Dox-40、ARP-1、ANBL-6 细胞 测试浓度:0.5、1、3、5、10 μM 孵育时间:24 小时 实验结果:所有细胞系均观察到生存力显着的浓度依赖性下降。 |
| 动物实验 |
Animal/Disease Models: CB-17 SCID mouse (human plasmacytoma MM.1S xenograft mouse model) [1]
Doses: 3 mg/kg Route of Administration: intravenous (iv) (iv)injection; twice a week for two weeks Experimental Results: Significant Dramatically inhibits MM tumor growth and prolongs mouse survival. |
| 药代性质 (ADME/PK) |
Absorption, Distribution and Excretion
For a 40 mg intravenous infusion, the active metabolite reaches a Cmax of 432 ng/mL, with a Tmax of 4-15 minutes, and an AUC of 3143 h\*µg/mL. Data regarding the route of elimination of melphalan flufenamide are not readily available. Free melphalan undergoes rapid and spontaneous decomposition, complicating studies on the route of elimination. However, it is expected to be mainly renally excreted. The mean volume of distribution of melphalan flufenamide is 35 L and of melphalan is 76 L. The mean clearance of melphalan flufenamide is 692 L/h and of melphalan is 23 L/h. Metabolism / Metabolites Melphalan flufenamide is metabolised to desethyl-melphalan and melphalan. Melphalan is spontaneously hydrolyzed to monohydroxy-melphalan and dihydroxy-melphalan. Biological Half-Life The mean elimination half life of melphalan flufenamide is 2.1 minutes and of melphalan is 70 minutes. |
| 毒性/毒理 (Toxicokinetics/TK) |
Protein Binding
Data regarding the protein binding of melphalan flufenamide are not readily available. However, free melphalan is 60% bound to albumin, 20% bound to alpha-1-acid glycoprotein, and 10% bound to other proteins in plasma. |
| 参考文献 | |
| 其他信息 |
Melphalan flufenamide, also known as melflufen or J1, is a prodrug of [melphalan]. Melphalan flufenamide is more readily uptaken by cells than melphalan, and is cleaved to the active metabolite by aminopeptidases. In vitro models show that melphalan is 10 to hundreds of times more potent than melphalan. The increased potency makes melphalan flufenamide a treatment option for patients with relapsed or refractory multiple myeloma who have attempted at least 4 lines of therapy already. Melphalan flufenamide was granted FDA approval on 26 February 2021.. It has since been withdrawn from the market in the wake of the phase 3 OCEAN trial which showed a decrease in overall survival in comparison to standard treatment with [pomalidomide] and [dexamethasone] despite superior progression-free survival.
Melphalan flufenamide is an Alkylating Drug. The mechanism of action of melphalan flufenamide is as an Alkylating Activity. Melphalan Flufenamide is a peptide-drug conjugate composed of a peptide conjugated, via an aminopeptidase-targeting linkage, to the alkylating agent melphalan, with potential antineoplastic and anti-angiogenic activities. Upon administration, the highly lipophilic melphalan flufenamide penetrates cell membranes and enters cells. In aminopeptidase-positive tumor cells, melphalan flufenamide is hydrolyzed by peptidases to release the hydrophilic alkylating agent melphalan. This results in the specific release and accumulation of melphalan in aminopeptidase-positive tumor cells. Melphalan alkylates DNA at the N7 position of guanine residues and induces DNA intra- and inter-strand cross-linkages. This results in the inhibition of DNA and RNA synthesis and the induction of apoptosis, thereby inhibiting tumor cell proliferation. Peptidases are overexpressed by certain cancer cells. The administration of melphalan flufenamide allows for enhanced efficacy and reduced toxicity compared to melphalan. See also: Melphalan Flufenamide Hydrochloride (annotation moved to). Drug Indication Melphalan flufenamide is indicated in combination with [dexamethasone] to treat adults with relapsed or refractory multiple myeloma who have received ≥4 therapies and are refractory to at least one proteasome inhibitor, immunomodulatory agent, and anti-CD38 monoclonal antibody. The FDA has withdrawn the drug from the market for this indication following phase 3 trial data showing decreased overall survival. Mechanism of Action Melphalan flufenamide is a more lipophilic prodrug of melphalan, which allows it to be more readily uptaken by cells. It is likely taken up into malignant cells by passive diffusion, where it is hydrolyzed by aminopeptidase N. The expression of aminopeptidases, along with other hydrolytic enzymes, is upregulated in many malignant cells, making the hydrolysis reaction to melphalan more favourable in a malignant cell. Increased concentrations of free melphalan in malignant cells leads to rapid irreversible DNA damage and apoptosis, reducing the potential for the development of resistance. Free melphalan is an nitrogen mustard derivative alkylating agent. Melphalan attaches alkyl groups to the N-7 position of guanine and N-3 position of adenine, leading to the formation of monoadducts, and DNA fragmenting when repair enzymes attempt to correct the error. It can also cause DNA cross-linking from the N-7 position of one guanine to the N-7 position of another, preventing DNA strands from separating for synthesis or transcription. Finally, melphalan can induce a number of different mutations. While melphalan induces phosphorylation of the DNA damage marker γ-H2AX in melphalan sensitive cells at 6 hours, melphalan flufenamide induces γ-H2AX at 2 hours. Melphalan flufenamide is also able to induce γ-H2AX in melphalan-resistant cells. Pharmacodynamics Melphalan flufenamide is an alkylating agent indicated to treat relapsed or refractory multiple myeloma in Melphalan flufenamide has a long duration of action as it is given every 28 days. Patients should be counselled regarding risks of thrombocytopenia, neutropenia, anemia, infections, secondary malignancies, embryo-fetal toxicity. |
| 分子式 |
C24H30CL2FN3O3
|
|---|---|
| 分子量 |
498.4204
|
| 精确质量 |
497.165
|
| CAS号 |
380449-51-4
|
| 相关CAS号 |
Melflufen hydrochloride;380449-54-7
|
| PubChem CID |
9935639
|
| 外观&性状 |
Typically exists as solid at room temperature
|
| LogP |
4.81
|
| tPSA |
88.15
|
| 氢键供体(HBD)数目 |
2
|
| 氢键受体(HBA)数目 |
6
|
| 可旋转键数目(RBC) |
14
|
| 重原子数目 |
33
|
| 分子复杂度/Complexity |
579
|
| 定义原子立体中心数目 |
2
|
| SMILES |
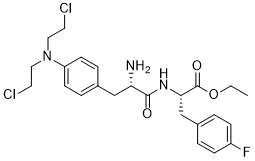
CCOC(=O)[C@H](CC1=CC=C(C=C1)F)NC(=O)[C@H](CC2=CC=C(C=C2)N(CCCl)CCCl)N
|
| InChi Key |
YQZNKYXGZSVEHI-VXKWHMMOSA-N
|
| InChi Code |
InChI=1S/C24H30Cl2FN3O3/c1-2-33-24(32)22(16-18-3-7-19(27)8-4-18)29-23(31)21(28)15-17-5-9-20(10-6-17)30(13-11-25)14-12-26/h3-10,21-22H,2,11-16,28H2,1H3,(H,29,31)/t21-,22-/m0/s1
|
| 化学名 |
ethyl (2S)-2-[[(2S)-2-amino-3-[4-[bis(2-chloroethyl)amino]phenyl]propanoyl]amino]-3-(4-fluorophenyl)propanoate
|
| 别名 |
J1 J-1 Prodrug J-1 MelflufenPepaxto
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples
|
|---|---|
| 溶解度 (体内实验) |
注意: 如下所列的是一些常用的体内动物实验溶解配方,主要用于溶解难溶或不溶于水的产品(水溶度<1 mg/mL)。 建议您先取少量样品进行尝试,如该配方可行,再根据实验需求增加样品量。
注射用配方
注射用配方1: DMSO : Tween 80: Saline = 10 : 5 : 85 (如: 100 μL DMSO → 50 μL Tween 80 → 850 μL Saline)(IP/IV/IM/SC等) *生理盐水/Saline的制备:将0.9g氯化钠/NaCl溶解在100 mL ddH ₂ O中,得到澄清溶液。 注射用配方 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (如: 100 μL DMSO → 400 μL PEG300 → 50 μL Tween 80 → 450 μL Saline) 注射用配方 3: DMSO : Corn oil = 10 : 90 (如: 100 μL DMSO → 900 μL Corn oil) 示例: 以注射用配方 3 (DMSO : Corn oil = 10 : 90) 为例说明, 如果要配制 1 mL 2.5 mg/mL的工作液, 您可以取 100 μL 25 mg/mL 澄清的 DMSO 储备液,加到 900 μL Corn oil/玉米油中, 混合均匀。 View More
注射用配方 4: DMSO : 20% SBE-β-CD in Saline = 10 : 90 [如:100 μL DMSO → 900 μL (20% SBE-β-CD in Saline)] 口服配方
口服配方 1: 悬浮于0.5% CMC Na (羧甲基纤维素钠) 口服配方 2: 悬浮于0.5% Carboxymethyl cellulose (羧甲基纤维素) 示例: 以口服配方 1 (悬浮于 0.5% CMC Na)为例说明, 如果要配制 100 mL 2.5 mg/mL 的工作液, 您可以先取0.5g CMC Na并将其溶解于100mL ddH2O中,得到0.5%CMC-Na澄清溶液;然后将250 mg待测化合物加到100 mL前述 0.5%CMC Na溶液中,得到悬浮液。 View More
口服配方 3: 溶解于 PEG400 (聚乙二醇400) 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.0063 mL | 10.0317 mL | 20.0634 mL | |
| 5 mM | 0.4013 mL | 2.0063 mL | 4.0127 mL | |
| 10 mM | 0.2006 mL | 1.0032 mL | 2.0063 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
A Study of Melphalan Flufenamide (Melflufen) Plus Dexamethasone in Patients With Relapsed or Refractory Multiple Myeloma
CTID: NCT02963493
Phase: Phase 2 Status: Completed
Date: 2022-11-22
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1

































