| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 10 mM * 1 mL in DMSO |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| 500mg |
|
||
| 1g |
|
||
| 2g |
|
||
| 5g |
|
||
| Other Sizes |
|

| 体外研究 (In Vitro) |
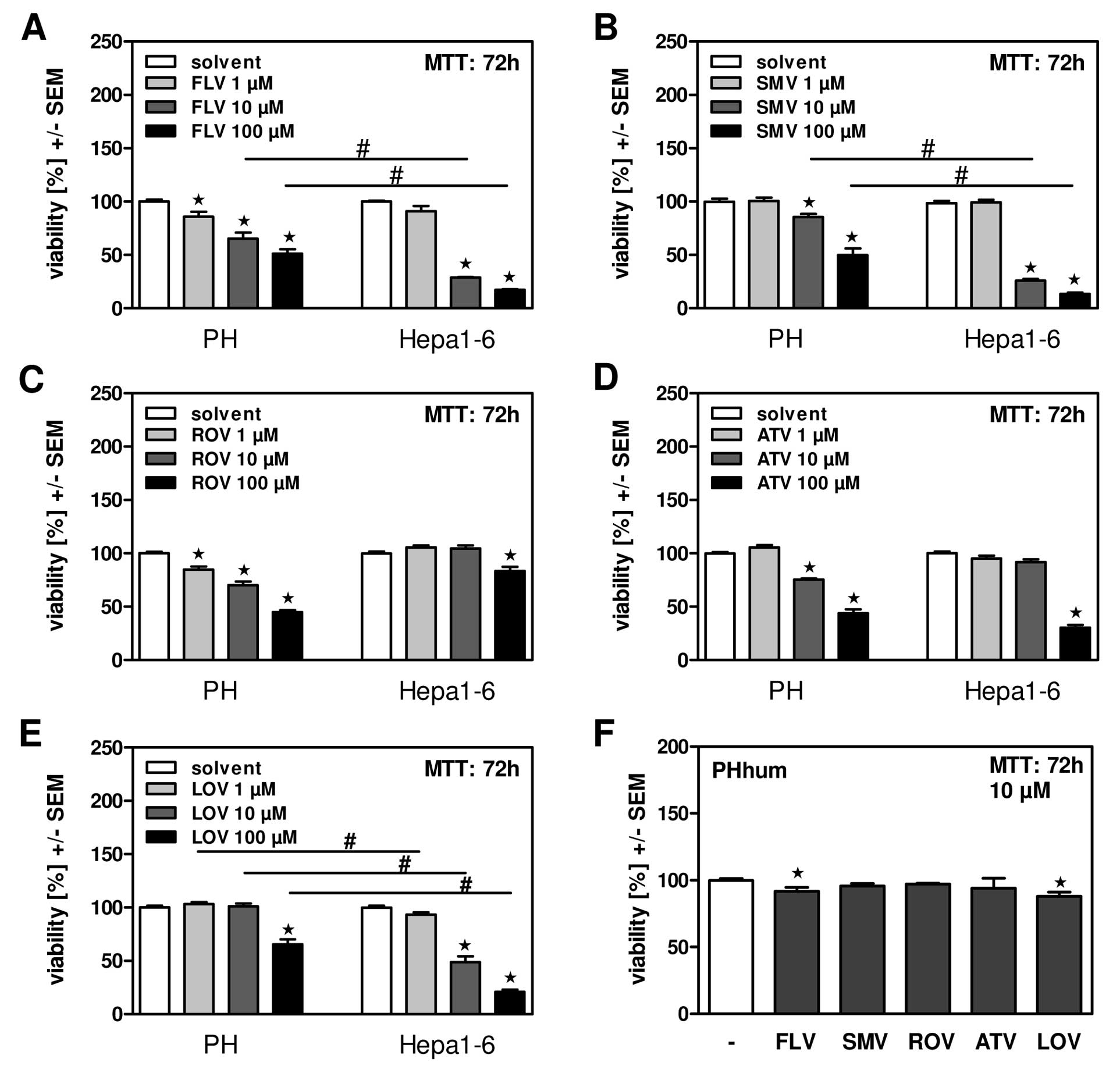
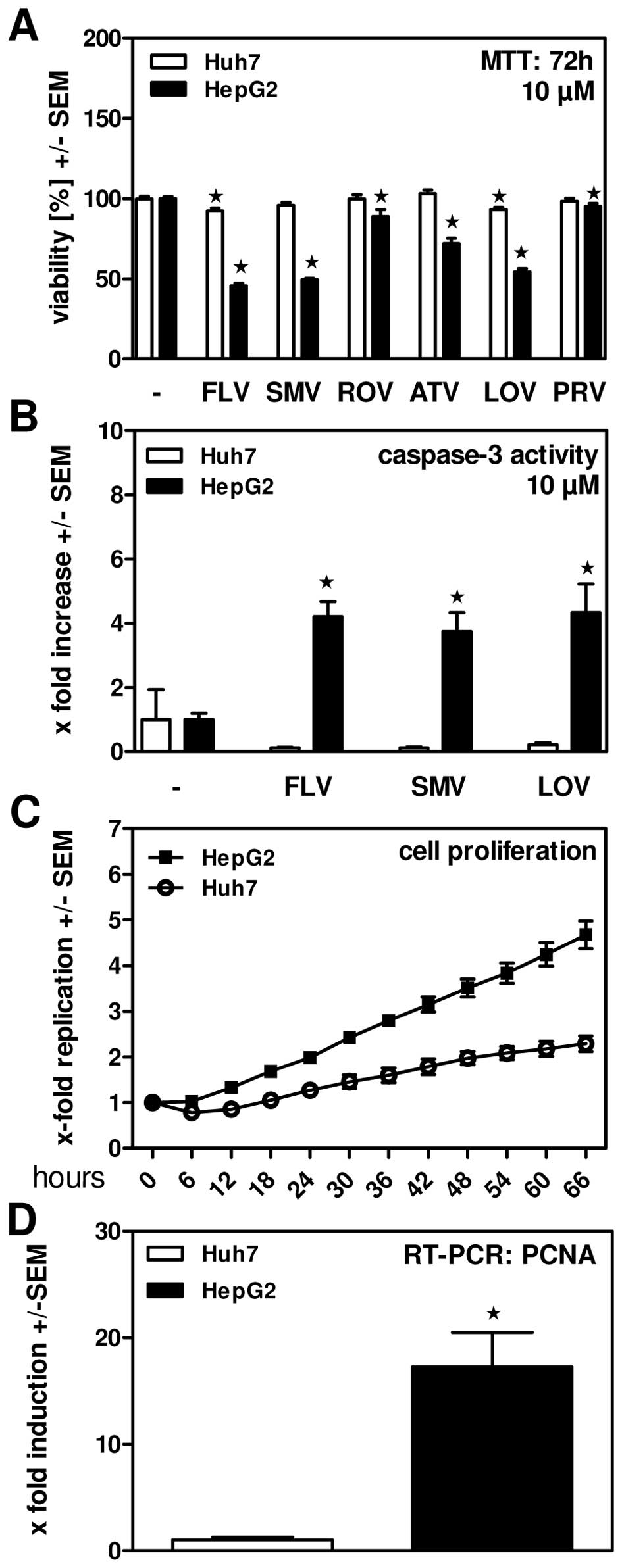
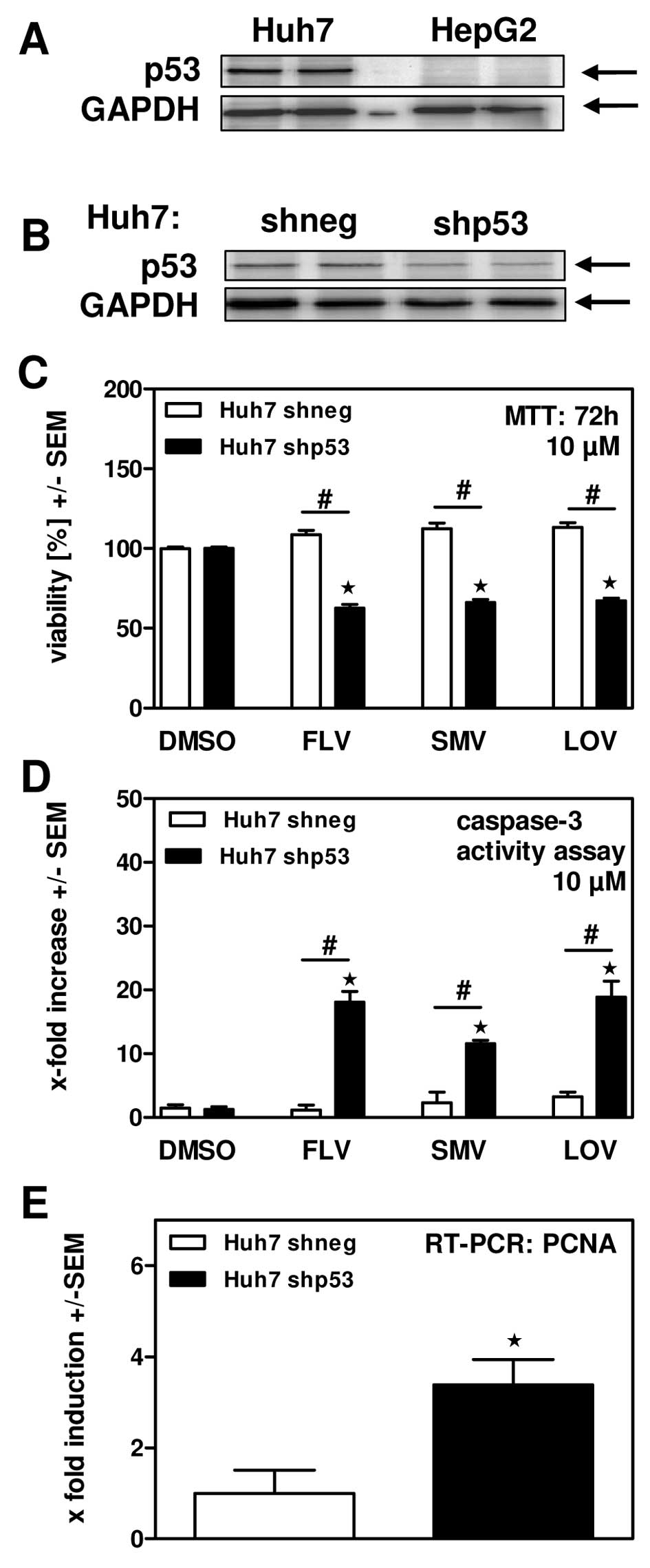
洛伐他汀(10 μM;72 小时)可有效降低 HepG2 细胞活力[2]。在 HepG2 细胞中,洛伐他汀(10 μM;48 小时)会导致细胞凋亡 [2]。
|
||
|---|---|---|---|
| 体内研究 (In Vivo) |
肝脏水解无活性的内酯洛伐他汀,产生活性的 f3-羟基酸形式。该主要代谢物抑制 HMG-CoA 还原酶。 Ki 是一纳米。人血浆蛋白与洛伐他汀及其β-羟基酸代谢物紧密结合。洛伐他汀可穿过胎盘屏障和血脑屏障[3]。洛伐他汀会适度升高 HDL 胆固醇,同时显着降低含载脂蛋白 B 的脂蛋白,特别是 LDL 胆固醇,并将血浆甘油三酯降至较低水平 [4]。
|
||
| 细胞实验 |
细胞活力测定[2]
细胞类型: HepG2 细胞 测试浓度: 10 μM 孵育时间: 72 hrs(小时) 实验结果:有效降低 HepG2 细胞的活力。 |
||
| 动物实验 |
|
||
| 药代性质 (ADME/PK) |
Absorption, Distribution and Excretion
Lovastatin Cmax was found to be 3.013ng/mL with a Tmax of 3.36 hours. Plasma concentrations of total radioactivity (lovastatin plus 14C-metabolites) peaked at 2 hours and declined rapidly to about 10% of peak by 24 hours postdose. Absorption of lovastatin, estimated relative to an intravenous reference dose, in each of four animal species tested, averaged about 30% of an oral dose. In animal studies, after oral dosing, lovastatin had high selectivity for the liver, where it achieved substantially higher concentrations than in non-target tissues. Lovastatin undergoes extensive first-pass extraction in the liver, its primary site of action, with subsequent excretion of drug equivalents in the bile. As a consequence of extensive hepatic extraction of lovastatin, the availability of drug to the general circulation is low and variable. In a single dose study in four hypercholesterolemic patients, it was estimated that less than 5% of an oral dose of lovastatin reaches the general circulation as active inhibitors. Following administration of lovastatin tablets the coefficient of variation, based on between-subject variability, was approximately 40% for the area under the curve (AUC) of total inhibitory activity in the general circulation. The peak concentrations of lovastatin when a dose of 10-40 mg is administered are reported to range from 1.04-4.03 ng/ml and an AUC of 14-53 ng.h/ml. This indicates that lovastatin presents a dose-dependent pharmacokinetic profile. When lovastatin was given under fasting conditions, plasma concentrations of both active and total inhibitors were on average about two-thirds those found when lovastatin was administered immediately after a standard test meal. Genetic differences in the OATP1B1 (Organic-Anion-Transporting Polypeptide 1B1) hepatic transporter encoded by the SCLCO1B1 gene (Solute Carrier Organic Anion Transporter family member 1B1) have been shown to impact lovastatin pharmacokinetics. Evidence from pharmacogenetic studies of the c.521T>C single nucleotide polymorphism (SNP) showed that lovastatin Cmax and AUC were 340 and 286% higher, respectively, for individuals homozygous for 521CC compared to homozygous 521TT individuals. The 521CC genotype is also associated with a marked increase in the risk of developing myopathy, likely secondary to increased systemic exposure. Other statin drugs impacted by this polymorphism include [rosuvastatin], [pitavastatin], [atorvastatin], [simvastatin], and [pravastatin]. While specific dosage instructions are not included in the available product monographs for lovastatin, individuals with the above-mentioned c.521CC OATP1B1 genotype should be monitored for development of adverse effects from increased exposure to the drug, such as muscle pain and risk of rhabdomyolysis, particularly at higher doses. Following an oral dose of 14C-labeled lovastatin to man, 10% of the dose was excreted in urine and 83% in feces. The latter represents absorbed drug excreted in bile, together with unabsorbed drug. Lovastatin is able to cross the blood-brain barrier and placenta. /MILK/ It is not known whether lovastatin is excreted in human milk. Following an oral dose of (14)C-labeled lovastatin in man, 10% of the dose was excreted in urine and 83% in feces. The latter represents absorbed drug equivalents excreted in bile, as well as any unabsorbed drug. Plasma concentrations of total radioactivity (lovastatin plus (14)C-metabolites) peaked at 2 hours and declined rapidly to about 10% of peak by 24 hours postdose. Absorption of lovastatin, estimated relative to an intravenous reference dose, in each of four animal species tested, averaged about 30% of an oral dose. In animal studies, after oral dosing, lovastatin had high selectivity for the liver, where it achieved substantially higher concentrations than in non-target tissues. Lovastatin undergoes extensive first-pass extraction in the liver, its primary site of action, with subsequent excretion of drug equivalents in the bile. As a consequence of extensive hepatic extraction of lovastatin, the availability of drug to the general circulation is low and variable. In a single dose study in four hypercholesterolemic patients, it was estimated that less than 5% of an oral dose of lovastatin reaches the general circulation as active inhibitors. Following administration of lovastatin tablets the coefficient of variation, based on between-subject variability, was approximately 40% for the area under the curve (AUC) of total inhibitory activity in the general circulation. Both lovastatin and its beta-hydroxyacid metabolite are highly bound (> 95%) to human plasma proteins. Animal studies demonstrated that lovastatin crosses the blood-brain and placental barriers. Peak plasma concentrations of both active and total inhibitors were attained within 2 to 4 hours of dose administration. Metabolism / Metabolites Lovastatin is given as a lactone prodrug and thus, in order to produce its mechanism of action, it is required to be converted to the active beta-hydroxy form. This drug activation process does not seem to be related to CYP isoenzyme activity but rather to be controlled by the activity of serum paraoxonase. Lovastatin is metabolized by the microsomal hepatic enzyme system (Cytochrome P-450 isoform 3A4). The major active metabolites present in human plasma are the β-hydroxy acid of lovastatin, its 6'-hydroxy, 6'-hydroxymethyl, and 6'-exomethylene derivatives. The uptake of lovastatin by the liver is enhanced by the activity of OATP1B1. Lovastatin is metabolized by the microsomal hepatic enzyme system (Cytochrome P-450 isoform 3A4). The major active metabolites present in human plasma are the beta-hydroxy acid of lovastatin, its 6'-hydroxy, 6'-hydroxymethyl, and 6'-exomethylene derivatives. The major active metabolites present in human plasma are the beta-hydroxyacid of lovastatin, its 6'-hydroxy derivative, and two additional metabolites. Lovastatin has known human metabolites that include 3-Hydroxylovastatin and 6'beta-Hydroxylovastatin. Lovastatin is hepatically metabolized in which the major active metabolites are the beta-hydroxyacid of lovastatin, the 6'-hydroxy derivative, and two additional metabolites. Route of Elimination: Lovastatin undergoes extensive first-pass extraction in the liver, its primary site of action, with subsequent excretion of drug equivalents in the bile. 83% of the orally administered dose is excreted in bile and 10% is excreted in urine. Half Life: 5.3 hours Biological Half-Life Lovastatin half-life is reported to be of 13.37 hours. The elimination half-life of the hydroxy acid form of lovastatin is reported to be of 0.7-3 hours. |
||
| 毒性/毒理 (Toxicokinetics/TK) |
Toxicity Summary
IDENTIFICATION AND USE: Lovastatin is a white, non-hygroscopic crystalline powder. It is used for therapy as anticholesteremic agent, and Hydroxymethylglutaryl-CoA (HMG-CoA) reductase inhibitor. HUMAN EXPOSURE AND TOXICITY: Lovastatin, like other inhibitors of HMG-CoA reductase, occasionally causes myopathy manifested as muscle pain, tenderness or weakness with creatine kinase above ten times the upper limit of normal. Myopathy sometimes takes the form of rhabdomyolysis with or without acute renal failure secondary to myoglobinuria, and rare fatalities have occurred. The risk of myopathy is increased by high levels of HMG-CoA reductase inhibitory activity in plasma. There have been rare postmarketing reports of fatal and non-fatal hepatic failure in patients taking statins, including lovastatin. Treatment of HL-60 cells with lovastatin induced characteristic apoptosis in a dose-dependent manner. ANIMAL STUDIES: Lovastatin produced optic nerve degeneration (Wallerian degeneration of retinogeniculate fibers) in clinically normal dogs in a dose-dependent fashion starting at 60 mg/kg/day. Vestibulocochlear Wallerian-like degeneration and retinal ganglion cell chromatolysis were also seen in dogs treated for 14 weeks at 180 mg/kg/day. CNS vascular lesions, characterized by perivascular hemorrhage and edema, mononuclear cell infiltration of perivascular spaces, perivascular fibrin deposits and necrosis of small vessels, were seen in dogs treated with lovastatin at a dose of 180 mg/kg/day. In mice given oral lovastatin dosages of 100 or 500 mg/kg daily, an increase in the incidence of papilloma in the nonglandular mucosa of the stomach was observed. Since the glandular mucosa of the stomach in these rodents was not affected and the human stomach contains only glandular mucosa, the importance of this finding to humans is unclear. An increased incidence of hepatocellular carcinoma and adenoma was observed after 21 months in mice given oral lovastatin 500 mg/kg daily. Drug-related testicular atrophy, decreased spermatogenesis, spermatocytic degeneration and giant cell formation were seen in dogs starting at 20 mg/kg/day. Direct dosing of neonatal rats by subcutaneous injection with 10 mg/kg/day of the open hydroxyacid form of lovastatin resulted in delayed passive avoidance learning in female rats. Lovastatin has been shown to produce skeletal malformations in offspring of pregnant mice and rats dosed during gestation at 80 mg/kg/day. Lovastatin did not exhibit mutagenic potential in in vitro mammalian cell systems (rat or mouse hepatocytes, V-79 cell forward mutation study), in vitro (Chinese hamster ovary cell) or in vivo (mouse bone marrow) chromosomal aberration studies, or microbial (Ames test) systems with or without metabolic activation. There is some in vitro evidence, however, that inhibition of HMG-CoA reductase can inhibit DNA synthesis during the S phase of the cell life cycle; this inhibition appears to result from depletion of mevalonic acid and is independent of its conversion to cholesterol. Lovastatin is structurally similar to the HMG, a substituent of the endogenous substrate of HMG-CoA reductase. Lovastatin is a prodrug that is activated in vivo via hydrolysis of the lactone ring to form the ‘_-hydroxyacid. The hydrolyzed lactone ring mimics the tetrahedral intermediate produced by the reductase allowing the agent to bind to HMG-CoA reductase with 20,000 times greater affinity than its natural substrate. The bicyclic portion of lovastatin binds to the coenzyme A portion of the active site. Hepatotoxicity Lovastatin therapy is associated with mild, asymptomatic and usually transient serum aminotransferase elevations. In summary analyses of large scale studies with prospective monitoring, ALT elevations above normal occurred in 3% to 5% of patients, but were above 3 times the upper limit of normal (ULN) in only 0.4% compared to 0.1% of placebo recipients. These elevations were more common with higher doses of lovastatin, being greater than 3 times ULN in 0.1% of patients receiving 20 mg daily, 0.9% with 40 mg and 1.5% with 80 mg daily. Most of these elevations were self-limited and did not require dose modification, although discontinuation is recommended for any elevation above 10 times and for persistent elevations above 5 times the ULN. Lovastatin is also associated with frank, clinically apparent hepatic injury, but cases are rare. The onset of clinical injury ranges from a few weeks to several years. The pattern of injury is typically cholestatic, but can be hepatocellular. Rash, fever and eosinophilia are uncommon as are autoimmune features. The injury usually resolves rapidly upon stopping lovastatin, but instances of fatal acute liver failure and of prolonged cholestasis have been reported (Case 1). The traditional Chinese medication known as red yeast rice which is used to treat hyperlipidemia has been shown to contain monacolin K, a natural component that is chemically identical to lovastatin, perhaps explaining its efficacy in reducing cholesterol levels. Red yeast rice has also been implicated in cases of acute liver injury and myopathies that are similar to those linked to lovastatin. In some instances cross-sensitivity to hepatic injury has been shown between red yeast rice products and lovastatin. Likelihood score: B (likely cause of clinically apparent liver injury). Effects During Pregnancy and Lactation ◉ Summary of Use during Lactation No relevant published information exists on the use of lovastatin during breastfeeding. Because of a concern with disruption of infant lipid metabolism, the consensus is that lovastatin should not be used during breastfeeding. However, others have argued that children homozygous for familial hypercholesterolemia are treated with statins beginning at 1 year of age, that statins have low oral bioavailability, and risks to the breastfed infant are low, especially with rosuvastatin and pravastatin.[1] Until more data become available, an alternate drug may be preferred, especially while nursing a newborn or preterm infant. ◉ Effects in Breastfed Infants Relevant published information was not found as of the revision date. ◉ Effects on Lactation and Breastmilk Relevant published information was not found as of the revision date. Protein Binding Both lovastatin and its β-hydroxy acid metabolite are highly bound (>95%) to human plasma proteins, largely due to its lipophilicity. Animal studies demonstrated that lovastatin crosses the blood-brain and placental barriers. Toxicity Data LD50>1000 mg/kg (orally in mice) Interactions ... A 60-year-old black man developed rhabdomyolysis after receiving lovastatin for 14 months. Rhabdomyolysis developed in the absence of other medications previously reported to cause this adverse effect when administered concomitantly with lovastatin. Adverse drug reaction causality algorithms categorized this reaction as either possible or probable. Rhabdomyolysis is an uncommon adverse effect associated with lovastatin therapy. Although reported cases of lovastatin-induced rhabdomyolysis were associated with the coadministration of cyclosporine, erythromycin, gemfibrozil, or nicotinic acid, this adverse effect may occur in the absence of these agents. Combined drug therapy should be approached with caution as information from controlled studies is limited. Based on post-marketing surveillance, gemfibrozil, other fibrates and lipid lowering doses of niacin (nicotinic acid) may increase the risk of myopathy when given concomitantly with HMG-CoA reductase inhibitors, probably because they can produce myopathy when given alone. Therefore, combined drug therapy should be approached with caution. Grapefruit juice contains one or more components that inhibit CYP3A4 and can increase the plasma levels of drugs metabolized by CYP3A4. The effect of typical consumption (one 250-ml glass daily) is minimal (34% increase in active plasma HMG-CoA reductase inhibitory activity as measured by the area under the concentration-time curve) and of no clinical relevance. However, because larger quantities (over 1 liter daily) significantly increase the plasma levels of HMG-CoA reductase inhibitory activity, grapefruit juice should be avoided during lovastatin therapy. Lovastatin, like several other inhibitors of HMG-CoA reductase, is a substrate of cytochrome P450 3A4 (CYP3A4). Certain drugs which inhibit this metabolic pathway can raise the plasma levels of lovastatin and may increase the risk of myopathy. These include itraconazole, ketoconazole, posaconazole, voriconazole, the macrolide antibiotics erythromycin and clarithromycin, the ketolide antibiotic telithromycin, HIV protease inhibitors, boceprevir, telaprevir, the antidepressant nefazodone, or cobicistat-containing products. Combination of these drugs with lovastatin is contraindicated. If short-term treatment with strong CYP3A4 inhibitors is unavoidable, therapy with lovastatin should be suspended during the course of treatment For more Interactions (Complete) data for Lovastatin (20 total), please visit the HSDB record page. Non-Human Toxicity Values LD50 Rat oral > 5000 mg/kg LD50 Mouse oral > 20,000 mg/kg |
||
| 参考文献 | |||
| 其他信息 |
Therapeutic Uses
Anticholesteremic Agents; Hydroxymethylglutaryl-CoA Reductase Inhibitors /CLINICAL TRIALS/ ClinicalTrials.gov is a registry and results database of publicly and privately supported clinical studies of human participants conducted around the world. The Web site is maintained by the National Library of Medicine (NLM) and the National Institutes of Health (NIH). Each ClinicalTrials.gov record presents summary information about a study protocol and includes the following: Disease or condition; Intervention (for example, the medical product, behavior, or procedure being studied); Title, description, and design of the study; Requirements for participation (eligibility criteria); Locations where the study is being conducted; Contact information for the study locations; and Links to relevant information on other health Web sites, such as NLM's MedlinePlus for patient health information and PubMed for citations and abstracts for scholarly articles in the field of medicine. Lovastatin is included in the database. Lovastatin Tablets USP are indicated as an adjunct to diet to reduce total cholesterol (total-C), low-density lipoprotein cholesterol (LDL-C) and apolipoprotein B levels in adolescent boys and girls who are at least one year post-menarche, 10 to 17 years of age, with heFH if after an adequate trial of diet therapy the following findings are present: 1. LDL-C remains > 189 mg/dL or 2. LDL-C remains > 160 mg/dL and: there is a positive family history of premature cardiovascular disease or two or more other CVD risk factors are present in the adolescent patient. /Included in US product label/ Therapy with lipid-altering agents should be a component of multiple risk factor intervention in those individuals at significantly increased risk for atherosclerotic vascular disease due to hypercholesterolemia. Lovastatin Tablets USP are indicated as an adjunct to diet for the reduction of elevated total cholesterol (total-C) and low-density lipoprotein cholesterol (LDL-C) levels in patients with primary hypercholesterolemia (Types IIa and IIb), when the response to diet restricted in saturated fat and cholesterol and to other nonpharmacological measures alone has been inadequate. /Included in US product label/ For more Therapeutic Uses (Complete) data for Lovastatin (9 total), please visit the HSDB record page. Drug Warnings Lovastatin, like other inhibitors of HMG-CoA reductase, occasionally causes myopathy manifested as muscle pain, tenderness or weakness with creatine kinase (CK) above ten times the upper limit of normal (ULN). Myopathy sometimes takes the form of rhabdomyolysis with or without acute renal failure secondary to myoglobinuria, and rare fatalities have occurred. The risk of myopathy is increased by high levels of HMG-CoA reductase inhibitory activity in plasma. Lovastatin is contraindicated in women who are or may become pregnant. The drug should be administered to women of childbearing age only when such patients are highly unlikely to conceive and have been informed of the potential hazard. If the patient becomes pregnant while receiving lovastatin, the drug should be discontinued immediately and the patient informed of the potential hazard to the fetus. Maternal treatment with lovastatin may reduce the fetal levels of mevalonate, which is a precursor of cholesterol biosynthesis. Atherosclerosis is a chronic process, and ordinarily discontinuation of lipid-lowering drugs during pregnancy should have little impact on the long-term risk associated with primary hypercholesterolemia. For these reasons, lovastatin should not be used in women who are pregnant, or can become pregnant. Lovastatin should be administered to women of child-bearing potential only when such patients are highly unlikely to conceive and have been informed of the potential hazards. Treatment should be immediately discontinued as soon as pregnancy is recognized. There have been rare postmarketing reports of fatal and non-fatal hepatic failure in patients taking statins, including lovastatin. If serious liver injury with clinical symptoms and/or hyperbilirubinemia or jaundice occurs during treatment with lovastatin, promptly interrupt therapy. If an alternate etiology is not found, do not restart lovastatin. For more Drug Warnings (Complete) data for Lovastatin (30 total), please visit the HSDB record page. Pharmacodynamics Lovastatin is an oral antilipemic agent which reversibly inhibits HMG-CoA reductase. It is used to lower total cholesterol, low density lipoprotein-cholesterol (LDL-C), apolipoprotein B (apoB), non-high density lipoprotein-cholesterol (non-HDL-C), and trigleride (TG) plasma concentrations while increasing HDL-C concentrations. High LDL-C, low HDL-C and high TG concentrations in the plasma are associated with increased risk of atherosclerosis and cardiovascular disease. The total cholesterol to HDL-C ratio is a strong predictor of coronary artery disease and high ratios are associated with higher risk of disease. Increased levels of HDL-C are associated with lower cardiovascular risk. By decreasing LDL-C and TG and increasing HDL-C, lovastatin reduces the risk of cardiovascular morbidity and mortality. Elevated cholesterol levels, and in particular, elevated low-density lipoprotein (LDL) levels, are an important risk factor for the development of CVD. Use of statins to target and reduce LDL levels has been shown in a number of landmark studies to significantly reduce the risk of development of CVD and all-cause mortality. Statins are considered a cost-effective treatment option for CVD due to their evidence of reducing all-cause mortality including fatal and non-fatal CVD as well as the need for surgical revascularization or angioplasty following a heart attack. Evidence has shown that even for low-risk individuals (with <10% risk of a major vascular event occurring within 5 years) statins cause a 20%-22% relative reduction in major cardiovascular events (heart attack, stroke, coronary revascularization, and coronary death) for every 1 mmol/L reduction in LDL without any significant side effects or risks. Clinical studies have shown that lovastatin reduces LDL-C and total cholesterol by 25-40%. The 50% inhibitory dose is known to be of 46 mcg/kg which is translated into a reduction of approximately 30% of plasma cholesterol. **Myopathy/Rhabdomyolysis** Lovastatin, like other inhibitors of HMG-CoA reductase, occasionally causes myopathy manifested as muscle pain, tenderness or weakness with creatine kinase (CK) above ten times the upper limit of normal (ULN). Myopathy sometimes takes the form of rhabdomyolysis with or without acute renal failure secondary to myoglobinuria, and rare fatalities have occurred. The risk of myopathy is dose-related and is increased by high levels of HMG-CoA reductase inhibitory activity in plasma. In a clinical study (EXCEL) in which patients were carefully monitored and some interacting drugs were excluded, there was one case of myopathy among 4933 patients randomized to lovastatin 20 to 40 mg daily for 48 weeks, and 4 among 1649 patients randomized to 80 mg daily. Predisposing factors for myopathy include advanced age (≥65 years), female gender, uncontrolled hypothyroidism, and renal impairment. Chinese patients may also be at increased risk for myopathy. In most cases, muscle symptoms and CK increases resolved when treatment was promptly discontinued. The risk of myopathy during treatment with lovastatin may be increased with concurrent administration of interacting drugs such as [fenofibrate], [niacin], [gemfibrozil], [cyclosporine], and strong inhibitors of the CYP3A4 enzyme. Cases of myopathy, including rhabdomyolysis, have been reported with HMG-CoA reductase inhibitors coadministered with [colchicine], and caution should therefore be exercised when prescribing these two medications together. Real-world data from observational studies has suggested that 10-15% of people taking statins may experience muscle aches at some point during treatment. **Liver Dysfunction** Persistent increases (to more than 3 times the upper limit of normal) in serum transaminases occurred in 1.9% of adult patients who received lovastatin for at least one year in early clinical trials. When the drug was interrupted or discontinued in these patients, the transaminase levels usually fell slowly to pretreatment levels. The increases usually appeared 3 to 12 months after the start of therapy with lovastatin, and were not associated with jaundice or other clinical signs or symptoms. In the EXCEL study, the incidence of persistent increases in serum transaminases over 48 weeks was 0.1% for placebo, 0.1% at 20 mg/day, 0.9% at 40 mg/day, and 1.5% at 80 mg/day in patients on lovastatin. However, in post-marketing experience with lovastatin, symptomatic liver disease has been reported rarely at all dosages. |
| 分子式 |
C24H36O5
|
|
|---|---|---|
| 分子量 |
404.54
|
|
| 精确质量 |
404.256
|
|
| CAS号 |
75330-75-5
|
|
| 相关CAS号 |
Lovastatin (Standard);75330-75-5;Lovastatin-d9;Lovastatin-d3;1002345-93-8
|
|
| PubChem CID |
53232
|
|
| 外观&性状 |
White to off-white solid powder
|
|
| 密度 |
1.1±0.1 g/cm3
|
|
| 沸点 |
559.2±50.0 °C at 760 mmHg
|
|
| 熔点 |
175°C
|
|
| 闪点 |
185.3±23.6 °C
|
|
| 蒸汽压 |
0.0±3.4 mmHg at 25°C
|
|
| 折射率 |
1.532
|
|
| LogP |
4.07
|
|
| tPSA |
72.83
|
|
| 氢键供体(HBD)数目 |
1
|
|
| 氢键受体(HBA)数目 |
5
|
|
| 可旋转键数目(RBC) |
7
|
|
| 重原子数目 |
29
|
|
| 分子复杂度/Complexity |
666
|
|
| 定义原子立体中心数目 |
8
|
|
| SMILES |
CC[C@H](C)C(=O)O[C@H]1C[C@H](C=C2[C@H]1[C@H]([C@H](C=C2)C)CC[C@@H]3C[C@H](CC(=O)O3)O)C
|
|
| InChi Key |
PCZOHLXUXFIOCF-BXMDZJJMSA-N
|
|
| InChi Code |
InChI=1S/C24H36O5/c1-5-15(3)24(27)29-21-11-14(2)10-17-7-6-16(4)20(23(17)21)9-8-19-12-18(25)13-22(26)28-19/h6-7,10,14-16,18-21,23,25H,5,8-9,11-13H2,1-4H3/t14-,15-,16-,18+,19+,20-,21-,23-/m0/s1
|
|
| 化学名 |
[(1S,3R,7S,8S,8aR)-8-[2-[(2R,4R)-4-hydroxy-6-oxooxan-2-yl]ethyl]-3,7-dimethyl-1,2,3,7,8,8a-hexahydronaphthalen-1-yl] (2S)-2-methylbutanoate
|
|
| 别名 |
|
|
| HS Tariff Code |
2934.99.9001
|
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
|
|||
|---|---|---|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 2.5 mg/mL (6.18 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 25.0 mg/mL澄清DMSO储备液加入到400 μL PEG300中,混匀;然后向上述溶液中加入50 μL Tween-80,混匀;加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: ≥ 2.5 mg/mL (6.18 mM) (饱和度未知) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 25.0 mg/mL澄清DMSO储备液加入900 μL 20% SBE-β-CD生理盐水溶液中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 View More
配方 3 中的溶解度: ≥ 2.5 mg/mL (6.18 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 配方 4 中的溶解度: 30% PEG400+0.5% Tween80+5% propylene glycol:30 mg/mL 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.4719 mL | 12.3597 mL | 24.7194 mL | |
| 5 mM | 0.4944 mL | 2.4719 mL | 4.9439 mL | |
| 10 mM | 0.2472 mL | 1.2360 mL | 2.4719 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
| NCT Number | Recruitment | interventions | Conditions | Sponsor/Collaborators | Start Date | Phases |
| NCT04297033 | Not yet recruiting | Drug: Lovastatin Drug: Placebo |
Cerebral Arteriovenous Malformation | Beijing Tiantan Hospital | January 1, 2021 | Phase 2 |
| NCT01527669 | Completed | Drug: LipoCol Forte capsules Drug: Lovastatin Tablet |
Healthy Subjects | National Taiwan University Hospital | February 2012 | Phase 4 |
| NCT00585052 | Terminated Has Results | Drug: Paclitaxel Drug: Lovastatin |
Ovarian Cancer | University of Iowa | August 2003 | Phase 2 |
| NCT01346670 | Completed | Drug: LipoCol and Mevacor | Healthy Volunteer | Taipei Medical University WanFang Hospital |
October 2006 | Phase 4 |
|
|
|
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1





























 NMR
NMR