| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 500mg |
|
||
| 1g |
|
||
| 2g |
|
||
| 5g |
|
||
| 10g |
|
||
| 50g |
|
||
| 100g |
|
||
| Other Sizes |
|

| 靶点 |
Quinolone; TOPO IV
|
|---|---|
| 体外研究 (In Vitro) |
体外活性:左氧氟沙星对大多数需氧革兰氏阳性菌和革兰氏阴性菌具有活性,对厌氧菌具有中等活性。左氧氟沙星对肺炎链球菌的活性比环丙沙星高两倍,对金黄色葡萄球菌、嗜麦芽黄单胞菌和脆弱拟杆菌的活性比环丙沙星高 2 至 4 倍。左氧氟沙星对凝固酶阴性葡萄球菌和不动杆菌的活性比环丙沙星高两到八倍,尽管这些效力的改善可能与临床无关。当使用浓度为 1 mg/mL 至 2 mg/mL 时,左氧氟沙星可抑制 90% 的链球菌。左氧氟沙星对细胞外或细胞内结核杆菌的抑制和杀菌活性是氧氟沙星的两倍。左氧氟沙星对成骨细胞生长的抑制作用最小,48和72小时时50%抑制浓度约为80 mg/mL。第 14 天通过茜素红染色和生化分析确定,左氧氟沙星可强烈抑制钙沉积。在培养的兔软骨细胞中,左氧氟沙星在实际关节病变浓度下首先抑制糖胺聚糖合成,其次抑制DNA合成和线粒体功能,但这些变化是可逆的,不足以杀死细胞。
|
| 体内研究 (In Vivo) |
对于小鼠全身感染和肾盂肾炎感染,左氧氟沙星与环丙沙星一样有效,甚至更有效。左氧氟沙星在小鼠血清和组织中的浓度高于环丙沙星。
|
| 动物实验 |
Matured male Albino mice
10.7 mg/kg Intraperitoneal injection; 10.7 mg/kg, once daily for 10 days or 3 weeks. |
| 药代性质 (ADME/PK) |
Absorption, Distribution and Excretion
Absorption of levofloxacin following oral administration is rapid and essentially complete, with an oral bioavailability of approximately 99%. Due to its nearly complete absorption, the intravenous and oral formulations of levofloxacin may be interchangeable. The Tmax is generally attained 1-2 hours following administration and the Cmax is proportional to the given dose - an intravenous dose of 500mg infused over 60 minutes resulted in a Cmax of 6.2 ± 1.0 µg/mL whereas a 750mg dose infused over 90 minutes resulted in a Cmax of 11.5 ± 4.0 µg/mL. Oral administration with food prolongs the Tmax by approximately 1 hour and slightly decreases the Cmax, but these changes are not likely to be clinically significant. Systemic absorption following oral inhalation is approximately 50% lower than that observed following oral administration. The majority of administered levofloxacin is excreted unchanged in the urine. Following the administration of a single oral dose of levofloxacin, approximately 87% was eliminated unchanged in the urine within 48 hours and less than 4% was eliminated in the feces within 72 hours. Levofloxacin is widely distributed in the body, with an average volume of distribution following oral administration between 1.09-1.26 L/kg (~89-112 L). Concentrations in many tissues and fluids may exceed those observed in plasma. Levofloxacin is known to penetrate well into skin tissue, fluids (e.g. blisters), lung tissue, and prostatic tissue, amongst others. The average apparent total body clearance of levofloxacin ranges from 8.64-13.56 L/h, and its renal clearance ranges from 5.76-8.52 L/h. The relative similarity of these ranges indicates a small degree of non-renal clearance. The mean volume of distribution of levofloxacin generally ranges from 74 to 112 L after single and multiple 500 mg or 750 mg doses, indicating widespread distribution into body tissues. Levofloxacin reaches its peak levels in skin tissues and in blister fluid of healthy subjects at approximately 3 hours after dosing. The skin tissue biopsy to plasma AUC ratio is approximately 2 and the blister fluid to plasma AUC ratio is approximately 1 following multiple once-daily oral administration of 750 mg and 500 mg doses of levaquin, respectively, to healthy subjects. Levofloxacin also penetrates well into lung tissues. Lung tissue concentrations were generally 2- to 5-fold higher than plasma concentrations and ranged from approximately 2.4 to 11.3 ug/g over a 24-hour period after a single 500 mg oral dose. Levofloxacin pharmacokinetics are linear and predictable after single and multiple oral or IV dosing regimens. Steady-state conditions are reached within 48 hours following a 500 mg or 750 mg once-daily dosage regimen. The mean + or - SD peak and trough plasma concentrations attained following multiple once-daily oral dosage regimens were approximately 5.7 + or - 1.4 and 0.5 + or - 0.2 ug/mL after the 500 mg doses, and 8.6+ or - 1.9 and 1.1 + or - 0.4 ug/mL after the 750 mg doses, respectively. The mean + or - SD peak and trough plasma concentrations attained following multiple once-daily IV regimens were approximately 6.4 + or - 0.8 and 0.6 + or - 0.2 ug/mL after the 500 mg doses, and 12.1+ or - 4.1 and 1.3 + or - 0.71 ug/mL after the 750 mg doses, respectively. Oral administration of a 500 mg dose of levaquin with food prolongs the time to peak concentration by approximately 1 hour and decreases the peak concentration by approximately 14% following tablet and approximately 25% following oral solution administration. Therefore, levaquin tablets can be administered without regard to food. It is recommended that levaquin oral solution be taken 1 hour before or 2 hours after eating. Levofloxacin is rapidly and essentially completely absorbed after oral administration. Peak plasma concentrations are usually attained one to two hours after oral dosing. The absolute bioavailability of levofloxacin from a 500 mg tablet and a 750 mg tablet of Levaquin are both approximately 99%, demonstrating complete oral absorption of levofloxacin. Following a single intravenous dose of Levaquin to healthy volunteers, the mean + or - SD peak plasma concentration attained was 6.2 + or - 1.0 ug/mL after a 500 mg dose infused over 60 minutes and 11.5+ or - 4.0 ug/mL after a 750 mg dose infused over 90 minutes. Levofloxacin is excreted largely as unchanged drug in the urine. The mean terminal plasma elimination half-life of levofloxacin ranges from approximately 6 to 8 hours following single or multiple doses of levofloxacin given orally or intravenously. The mean apparent total body clearance and renal clearance range from approximately 144 to 226 mL/min and 96 to 142 mL/min, respectively. Renal clearance in excess of the glomerular filtration rate suggests that tubular secretion of levofloxacin occurs in addition to its glomerular filtration. Concomitant administration of either cimetidine or probenecid results in approximately 24% and 35% reduction in the levofloxacin renal clearance, respectively, indicating that secretion of levofloxacin occurs in the renal proximal tubule. No levofloxacin crystals were found in any of the urine samples freshly collected from subjects receiving levaquin. For more Absorption, Distribution and Excretion (Complete) data for Levofloxacin (6 total), please visit the HSDB record page. Metabolism / Metabolites Only 2 metabolites, desmethyl-levofloxacin and levofloxacin-N-oxide, have been identified in humans, neither of which appears to carry any relevant pharmacological activity. Following oral administration, less than 5% of the administered dose was recovered in the urine as these metabolites, indicating very little metabolism of levofloxacin in humans. The specific enzymes responsible for the demethylation and oxidation of levofloxacin have yet to be ascertained. Levofloxacin is stereochemically stable in plasma and urine and does not invert metabolically to its enantiomer, D-ofloxacin. Levofloxacin undergoes limited metabolism in humans and is primarily excreted as unchanged drug in the urine. Following oral administration, approximately 87% of an administered dose was recovered as unchanged drug in urine within 48 hours, whereas less than 4% of the dose was recovered in feces in 72 hours. Less than 5% of an administered dose was recovered in the urine as the desmethyl and N-oxide metabolites, the only metabolites identified in humans. These metabolites have little relevant pharmacological activity. Mainly excreted as unchanged drug (87%); undergoes limited metabolism in humans. Levofloxacin is rapidly and essentially completely absorbed after oral administration. It is distributed into body tissues, especially in the skin and lung tissues. Levofloxacin is stereochemically stable in plasma and urine and does not invert metabolically to its enantiomer, D-ofloxacin. Levofloxacin undergoes limited metabolism in humans and is primarily excreted as unchanged drug in the urine. Following oral administration, approximately 87% of an administered dose was recovered as unchanged drug in urine within 48 hours, whereas less than 4% of the dose was recovered in feces in 72 hours. Less than 5% of an administered dose was recovered in the urine as the desmethyl and N-oxide metabolites, the only metabolites identified in humans. These metabolites have little relevant pharmacological activity. Levofloxacin is excreted largely as unchanged drug in the urine (L1009). Route of Elimination: Mainly excreted as unchanged drug in the urine. Half Life: 6-8 hours Biological Half-Life The average terminal elimination half-life of levofloxacin is 6-8 hours. The mean terminal plasma elimination half-life of levofloxacin ranges from approximately 6 to 8 hours following single or multiple doses of levofloxacin given orally or intravenously. The pharmacokinetics of oral levofloxacin and its penetration into inflammatory fluid were studied in 6 healthy male subjects (ages 18-45 yr) who received 500 mg drug every 12 hr for 5 doses or 500 mg every 24 hr for 3 doses in an open crossover design. ... Mean terminal elimination half-lives in plasma were 7.9 and 8 hr for the 2 regimens, respectively, and the same values were seen for inflammatory fluid. ... After single oral administration of (14)C-levofloxacin at a dose of 20 mg kg(-1) under non-fasting conditions, the absorption, distribution and excretion of radioactivity were studied in albino and pigmented rats. ... The uveal tract concentrations reached the maximum value (C(max)) of 26.33 +/- 0.75 ug eq. g(-1) at 24 hr after dosing and declined slowly with a terminal half-life of 468.1 hr (19.5 days). |
| 毒性/毒理 (Toxicokinetics/TK) |
Toxicity Summary
Levofloxacin inhibits bacterial type II topoisomerases, topoisomerase IV and DNA gyrase. Levofloxacin, like other fluoroquinolones, inhibits the A subunits of DNA gyrase, two subunits encoded by the gyrA gene. This results in strand breakage on a bacterial chromosome, supercoiling, and resealing; DNA replication and transcription is inhibited. Hepatotoxicity In short term studies, levofloxacin has been associated with minor elevations in serum ALT and AST levels in 2% to 5% of patients. The abnormalities were usually asymptomatic and transient and rarely require dose modification. With its wide scale use, levofloxacin has been implicated in in at least 50 instances of clinically apparent liver injury mostly in isolated case reports. The clinical presentation and course are typical of the hepatotoxicity of other fluoroquinolones, and the injury is likely a class effect. The latency to onset is usually short (1 to 3 weeks) and the onset is often abrupt with a hepatocellular or mixed pattern of injury, jaundice and, in some instances, hepatic failure. Cholestatic hepatitis can also occur. Immunoallergic features such as fever, rash and eosinophilia are common, but not particularly prominent. Autoantibodies are rare. The liver injury is usually self-limited, but several cases of acute liver failure have been linked to fluoroquinolones as well as instances of prolonged jaundice, cholestasis and vanishing bile duct syndrome. Levofloxacin, like ciprofloxacin, has also been implicated hypersensitivity reactions including rare cases of Stevens Johnson syndrome and toxic epidermal necrolysis, which may be accompanied by liver injury. While liver injury from levofloxacin is rare, the fluoroquinolones collectively are among the most frequent causes of clinically apparent liver injury including fatal cases and cases of chronic liver injury and bile duct paucity. Likelihood score: A (well established cause of clinically apparent liver injury). Effects During Pregnancy and Lactation ◉ Summary of Use during Lactation Levofloxacin is the S-enantiomer of the fluoroquinolone, ofloxacin. No information is available on the clinical use of levofloxacin during breastfeeding. However, amounts in breastmilk appear to be far lower than the infant dose and would not be expected to cause any adverse effects in breastfed infants. Fluoroquinolones such as levofloxacin have traditionally not been used in infants because of concern about adverse effects on the infants' developing joints. However, more recent studies indicate little risk. The calcium in milk might prevent absorption of the small amounts of fluoroquinolones in milk, but insufficient data exist to prove or disprove this assertion. Use of levofloxacin is acceptable in nursing mothers with monitoring of the infant for possible effects on the gastrointestinal flora, such as diarrhea or candidiasis (thrush, diaper rash). Avoiding breastfeeding for 4 to 6 hours after a dose should decrease the exposure of the infant to levofloxacin in breastmilk. Maternal use of an eye drop that contains levofloxacin presents negligible risk for the nursing infant. ◉ Effects in Breastfed Infants Relevant published information was not found as of the revision date. ◉ Effects on Lactation and Breastmilk Relevant published information was not found as of the revision date. Protein Binding Levofloxacin is 24-38% protein-bound in plasma, primarily to albumin. The extent of protein-binding is independent of its plasma concentration. Toxicity Data LD50: 640 mg/kg (Oral, Rat) Interactions Warfarin: Potential pharmacologic interaction (increased prothrombin time). Monitor prothrombin time or other suitable coagulation tests and monitor for bleeding. Theophylline: Pharmacokinetic interaction unlikely. However, pharmacokinetic interaction (increased theophylline half-life and increased risk of theophylline-related adverse effects) occurs with some other quinolones. Closely monitor serum theophylline concentrations and adjust theophylline dosage accordingly; consider that adverse theophylline effects (e.g., seizures) may occur with or without elevated theophylline concentrations. Sucralfate: Potential pharmacokinetic interaction (decreased levofloxacin absorption); no pharmacokinetic interaction if given 2 hours apart. Administer levofloxacin at least 2 hours before or 2 hours after sucralfate. Probenecid: Potential pharmacokinetic interaction (increased levofloxacin AUC and half-life). Not considered clinically important; dosage adjustments are not required. For more Interactions (Complete) data for Levofloxacin (16 total), please visit the HSDB record page. |
| 参考文献 | |
| 其他信息 |
Therapeutic Uses
Anti-Bacterial Agents; Anti-Infective Agents, Urinary; Nucleic Acid Synthesis Inhibitors Levofloxacin is used for the treatment of acute bacterial sinusitis caused by susceptible Streptococcus pneumoniae, Haemophilus influenzae, or Moraxella catarrhalis. /Included in US product label/ Levofloxacin is used for the treatment of community-acquired pneumonia caused by susceptible S. aureus (oxacillin-susceptible strains), S. pneumoniae (including penicillin-resistant strains (penicillin MIC of 2 ug/mL or greater)), H. influenzae, H. parainfluenzae, Klebsiella pneumoniae, Legionella pneumophila, M. catarrhalis, Chlamydophila pneumoniae (formerly Chlamydia pneumoniae), or Mycoplasma pneumoniae. /Included in US product label/ Levofloxacin is used for the treatment of mild to moderate complicated urinary tract infections caused by susceptible E. faecalis, Enterobacter cloacae, E. coli, K. pneumoniae, P. mirabilis, or Ps. aeruginosa and acute pyelonephritis caused by susceptible E. coli, including cases with concurrent bacteremia. /Included in US product label/ For more Therapeutic Uses (Complete) data for Levofloxacin (23 total), please visit the HSDB record page. Drug Warnings /BOXED WARNING/ WARNING: Fluoroquinolones, including Levaquin, are associated with an increased risk of tendinitis and tendon rupture in all ages. This risk is further increased in older patients usually over 60 years of age, in patients taking corticosteroid drugs, and in patients with kidney, heart or lung transplants. /BOXED WARNING/ WARNING: Fluoroquinolones, including Levaquin may exacerbate muscle weakness in persons with myasthenia gravis. Avoid Levaquin in patients with a known history of myasthenia gravis Other serious and sometimes fatal events, some due to hypersensitivity, and some due to uncertain etiology, have been reported rarely in patients receiving therapy with fluoroquinolones, including Levaquin. These events may be severe and generally occur following the administration of multiple doses. Clinical manifestations may include one or more of the following: fever, rash, or severe dermatologic reactions (e.g., toxic epidermal necrolysis, Stevens-Johnson Syndrome); vasculitis; arthralgia; myalgia; serum sickness; allergic pneumonitis; interstitial nephritis; acute renal insufficiency or failure; hepatitis; jaundice; acute hepatic necrosis or failure; anemia, including hemolytic and aplastic; thrombocytopenia, including thrombotic thrombocytopenic purpura; leukopenia; agranulocytosis; pancytopenia; and/or other hematologic abnormalities. The drug should be discontinued immediately at the first appearance of skin rash, jaundice, or any other sign of hypersensitivity and supportive measures instituted. Post-marketing reports of severe hepatotoxicity (including acute hepatitis and fatal events) have been received for patients treated with Levaquin No evidence of serious drug-associated hepatotoxicity was detected in clinical trials of over 7,000 patients. Severe hepatotoxicity generally occurred within 14 days of initiation of therapy and most cases occurred within 6 days. Most cases of severe hepatotoxicity were not associated with hypersensitivity. The majority of fatal hepatotoxicity reports occurred in patients 65 years of age or older and most were not associated with hypersensitivity. Levaquin should be discontinued immediately if the patient develops signs and symptoms of hepatitis. For more Drug Warnings (Complete) data for Levofloxacin (19 total), please visit the HSDB record page. Pharmacodynamics Levofloxacin is bactericidal and exerts its antimicrobial effects via inhibition of bacterial DNA replication. It has a relatively long duration of action in comparison with other antibiotics that allows for once or twice daily dosing. Levofloxacin is associated with QTc-interval prolongation and should be used with caution in patients with other risk factors for prolongation (e.g. hypokalemia, concomitant medications). Levofloxacin has demonstrated _in vitro_ activity against a number of aerobic gram-positive and gram-negative bacteria and may carry some activity against certain species of anaerobic bacteria and other pathogens such as _Chlamydia_ and _Legionella_. Resistance to levofloxacin may develop, and is generally due to mutations in DNA gyrase or topoisomerase IV, or via alterations to drug efflux. Cross-resistance may occur between levofloxacin and other fluoroquinolones, but is unlikely to develop between levofloxacin and other antibiotic classes (e.g. macrolides) due to significant differences in chemical structure and mechanism of action. As antimicrobial susceptibility patterns are geographically distinct, local antibiograms should be consulted to ensure adequate coverage of relevant pathogens prior to use. |
| 分子式 |
C18H20FN3O4
|
|
|---|---|---|
| 分子量 |
361.37
|
|
| 精确质量 |
361.143
|
|
| 元素分析 |
C, 59.83; H, 5.58; F, 5.26; N, 11.63; O, 17.71
|
|
| CAS号 |
100986-85-4
|
|
| 相关CAS号 |
138199-71-0; 177325-13-2;872606-49-0
|
|
| PubChem CID |
149096
|
|
| 外观&性状 |
Off-white to light yellow solid powder
|
|
| 密度 |
1.5±0.1 g/cm3
|
|
| 沸点 |
571.5±50.0 °C at 760 mmHg
|
|
| 熔点 |
218ºC
|
|
| 闪点 |
299.4±30.1 °C
|
|
| 蒸汽压 |
0.0±1.7 mmHg at 25°C
|
|
| 折射率 |
1.670
|
|
| LogP |
0.84
|
|
| tPSA |
75.01
|
|
| 氢键供体(HBD)数目 |
1
|
|
| 氢键受体(HBA)数目 |
8
|
|
| 可旋转键数目(RBC) |
2
|
|
| 重原子数目 |
26
|
|
| 分子复杂度/Complexity |
634
|
|
| 定义原子立体中心数目 |
1
|
|
| SMILES |
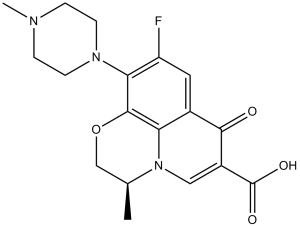
FC1C([H])=C2C(C(C(=O)O[H])=C([H])N3C2=C(C=1N1C([H])([H])C([H])([H])N(C([H])([H])[H])C([H])([H])C1([H])[H])OC([H])([H])[C@]3([H])C([H])([H])[H])=O
|
|
| InChi Key |
GSDSWSVVBLHKDQ-JTQLQIEISA-N
|
|
| InChi Code |
InChI=1S/C18H20FN3O4/c1-10-9-26-17-14-11(16(23)12(18(24)25)8-22(10)14)7-13(19)15(17)21-5-3-20(2)4-6-21/h7-8,10H,3-6,9H2,1-2H3,(H,24,25)/t10-/m0/s1
|
|
| 化学名 |
(2S)-7-fluoro-2-methyl-6-(4-methylpiperazin-1-yl)-10-oxo-4-oxa-1-azatricyclo[7.3.1.05,13]trideca-5(13),6,8,11-tetraene-11-carboxylic acid
|
|
| 别名 |
|
|
| HS Tariff Code |
2934.99.9001
|
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month 注意: 本产品在运输和储存过程中需避光。 |
|
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
|
|||
|---|---|---|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 1 mg/mL (2.77 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 10.0 mg/mL澄清DMSO储备液加入400 μL PEG300中,混匀;然后向上述溶液中加入50 μL Tween-80,混匀;加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: ≥ 1 mg/mL (2.77 mM) (饱和度未知) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 10.0 mg/mL澄清DMSO储备液加入900 μL 20% SBE-β-CD生理盐水溶液中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 View More
配方 3 中的溶解度: ≥ 1 mg/mL (2.77 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 配方 4 中的溶解度: 10 mg/mL (27.67 mM) in PBS (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液; 超声助溶. 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.7672 mL | 13.8362 mL | 27.6725 mL | |
| 5 mM | 0.5534 mL | 2.7672 mL | 5.5345 mL | |
| 10 mM | 0.2767 mL | 1.3836 mL | 2.7672 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
Effectiveness of Single Dose Fosfomycin and Single Dose Levofloxacin as Pre-urodynamic Antibiotic for UTI Prevention
CTID: NCT06017479
Phase: Phase 1/Phase 2 Status: Completed
Date: 2024-10-09
 SDOX
SDOX
 Topoisomerase I/II inhibitor 2
Topoisomerase I/II inhibitor 2
 Camptothecin-20(S)-O-propionate hydrate (Camptothecin-20-O-propionate hydrate)
Camptothecin-20(S)-O-propionate hydrate (Camptothecin-20-O-propionate hydrate)
 Topoisomerase I inhibitor 9
Topoisomerase I inhibitor 9
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1





























 COA
COA
