| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| 500mg |
|
||
| Other Sizes |
|
| 药代性质 (ADME/PK) |
Absorption, Distribution and Excretion
Bioavailablity after intramuscular injection of the depot formulation is estimated to be about 90%. The pharmacological effects of leuprolide acetate depot microspheres were studied in rats and dogs following subcutaneous and intramuscular injection. After injection the microspheres provided similar linear drug release and sustained serum drug levels for 3 months. Persistent suppression of serum luteinizing hormone, follicle stimulating hormone in rats, and testosterone in rats and dogs for over 16 wk was achieved with microspheres at a dose of 100 ug/kg/day in rats and 25.6 ug/kg/day in dogs. Responses upon periodic challenge tests revealed that a single injection of microspheres dramatically suppressed the function of the pituitary-gonadal system for 15 wks in rats. The growth of genital organs was also suppressed dose-dependently for over 3 months. It was concluded that persistent pharmacological effects are obtained with an injection of leuprolide 3-month depot microspheres. The effect of formulation adjuvants on the absorption of leuprolide acetate after intraduodenal injection and oral administration to male castrate rats is reported. Absorption was low, approximately 0.01% and 0.08% by oral and intraduodenal administration, respectively, compared with intravenous controls. An aqueous formulation and a water-in-oil emulsion of a lipophilic salt, a decane sulfonic acid derivative of leuprolide gave intraduodenal bioavailabilities of approximately 0.2% and 1% respectively. Evaluation of formulation effects on the oral absorption of the drug showed that lipophilicity, surfactant, and vehicle properties significantly affected intraduodenal absorption of leuprolide. Absolute bioavailability of the drug in typical emulsion systems ranged from approximately 3-10% and represented an improvement of about 100-fold in gastrointestinal bioavailability of this peptide. The implications of these findings relative to the effect of formulation adjuvants on oral absorption of leuprolide and other peptides following intraduodenal administration are discussed. The bioavailability of leuprolide acetate was studied in rats and in healthy males (ages 19-39 yr) after inhalation and intranasal administration, compared with intravenous and subcutaneous injection. Intranasal bioavailability in rats was significantly increased by alpha-cyclodextrin, eidetic acid, and solution volume. Intra-animal variability was 30-60% and absorption ranged from 8 to 46% compared with intravenous controls. In humans, the subcutaneous injection was 94% bioavailable compared with intravenous. Intranasal bioavailability averaged 2.4%, with significant intersubject variability. Plasma peak concentrations for one and 3 mg dosages were 0.24-1.6 and 0.1-11 ng/ml, respectively. Mean plasma peak concentrations of one mg aerosol and 2 mg suspension aerosols, respectively. Bioavailability of suspension aerosols was fourfold greater than that of the solution aerosol. /Leuprolide acetate/ |
|---|---|
| 其他信息 |
Leuprolide Acetate can cause developmental toxicity, female reproductive toxicity and male reproductive toxicity according to state or federal government labeling requirements.
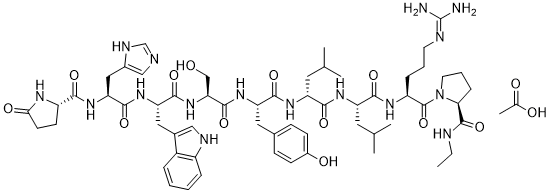
Leuprolide acetate is an acetate salt obtained by combining the nonapeptide leuprolide with acetic acid. A long lasting GnRH analog, LH-Rh agonist. It is a synthetic nonapeptide analogue of gonadotropin-releasing hormone, and is used as a subcutaneous hydrogel implant for the treatment of prostate cancer and for the suppression of gonadal sex hormone production in children with central precocious puberty. It has a role as an antineoplastic agent and a gonadotropin releasing hormone agonist. It contains a leuprolide. Leuprolide Acetate is the acetate salt of a synthetic nonapeptide analogue of gonadotropin-releasing hormone. Leuprolide binds to and activates gonadotropin-releasing hormone (GnRH) receptors. Continuous, prolonged administration of leuprolide in males results in pituitary GnRH receptor desensitization and inhibition of pituitary secretion of follicle stimulating hormone (FSH) and luteinizing hormone (LH), leading to a significant decline in testosterone production; in females, prolonged administration results in a decrease in estradiol production. This agent reduces testosterone production to castration levels and may inhibit androgen receptor-positive tumor progression. A potent synthetic long-acting agonist of GONADOTROPIN-RELEASING HORMONE that regulates the synthesis and release of pituitary gonadotropins, LUTEINIZING HORMONE and FOLLICLE STIMULATING HORMONE. See also: Leuprolide (has active moiety); Leuprolide acetate; norethindrone acetate (component of). Mechanism of Action Like naturally occurring luteinizing hormone-releasing hormone, initial or intermittent administration of leuprolide stimulates release of luteinizing hormone and follicle-stimulating hormone from the anterior pituitary. Luteinizing hormone and follicle-stimulating hormone release from the anterior pituitary transiently increases testosterone concentration in males. However, continuous administration of leuprolide in the treatment of prostatic carcinoma suppresses secretion of gonadotropin-releasing hormone, with a resultant fall in testosterone concentrations and a "medical castration". Initial stimulation of gonadotropins form the anterior pituitary is followed by prolonged suppression. Gonadotropin release from the anterior pituitary transiently increases estrone and estradiol concentrations in females. However, continuous administration of leuprolide in the treatment of endometriosis produces a fall in estrogens to postmenopausal levels. As a consequence of suppression of ovarian function, both normal and ectopic endometrial tissues become inactive and atrophic. As a result, amenorrhea occurs. |
| 分子式 |
C61H88N16O14
|
|---|---|
| 分子量 |
1269.473
|
| 精确质量 |
1268.666
|
| CAS号 |
74381-53-6
|
| 相关CAS号 |
53714-56-0 (Parent)
|
| PubChem CID |
657180
|
| 外观&性状 |
Fluffy solid
|
| 沸点 |
1720.5ºC at 760 mmHg
|
| 熔点 |
150-155ºC
|
| 闪点 |
994.3ºC
|
| LogP |
3.447
|
| tPSA |
466.34
|
| 氢键供体(HBD)数目 |
16
|
| 氢键受体(HBA)数目 |
16
|
| 可旋转键数目(RBC) |
32
|
| 重原子数目 |
91
|
| 分子复杂度/Complexity |
2420
|
| 定义原子立体中心数目 |
9
|
| SMILES |
O=C([C@]([H])(C([H])([H])C([H])([H])C([H])([H])/N=C(\N([H])[H])/N([H])[H])N([H])C([C@]([H])(C([H])([H])C([H])(C([H])([H])[H])C([H])([H])[H])N([H])C([C@@]([H])(C([H])([H])C([H])(C([H])([H])[H])C([H])([H])[H])N([H])C([C@]([H])(C([H])([H])C1C([H])=C([H])C(=C([H])C=1[H])O[H])N([H])C([C@]([H])(C([H])([H])O[H])N([H])C([C@]([H])(C([H])([H])C1=C([H])N([H])C2=C([H])C([H])=C([H])C([H])=C12)N([H])C([C@]([H])(C([H])([H])C1=C([H])N=C([H])N1[H])N([H])C([C@]1([H])C([H])([H])C([H])([H])C(N1[H])=O)=O)=O)=O)=O)=O)=O)=O)N1C([H])([H])C([H])([H])C([H])([H])[C@@]1([H])C(N([H])C([H])([H])C([H])([H])[H])=O.O([H])C(C([H])([H])[H])=O
|
| InChi Key |
RGLRXNKKBLIBQS-XNHQSDQCSA-N
|
| InChi Code |
InChI=1S/C59H84N16O12.C2H4O2/c1-6-63-57(86)48-14-10-22-75(48)58(87)41(13-9-21-64-59(60)61)68-51(80)42(23-32(2)3)69-52(81)43(24-33(4)5)70-53(82)44(25-34-15-17-37(77)18-16-34)71-56(85)47(30-76)74-54(83)45(26-35-28-65-39-12-8-7-11-38(35)39)72-55(84)46(27-36-29-62-31-66-36)73-50(79)40-19-20-49(78)67-401-2(3)4/h7-8,11-12,15-18,28-29,31-33,40-48,65,76-77H,6,9-10,13-14,19-27,30H2,1-5H3,(H,62,66)(H,63,86)(H,67,78)(H,68,80)(H,69,81)(H,70,82)(H,71,85)(H,72,84)(H,73,79)(H,74,83)(H4,60,61,64)1H3,(H,3,4)/t40-,41-,42-,43+,44-,45-,46-,47-,48-/m0./s1
|
| 化学名 |
Pyr-His-Trp-Ser-Tyr-D-Leu-Leu-Arg-Pro-NHEt acetate
|
| 别名 |
Abbott-43818 A-43818 ELIGARD Lupron LEUP A43818 TAP144Abbott 43818Lupron Depot
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples
|
|---|---|
| 溶解度 (体内实验) |
注意: 如下所列的是一些常用的体内动物实验溶解配方,主要用于溶解难溶或不溶于水的产品(水溶度<1 mg/mL)。 建议您先取少量样品进行尝试,如该配方可行,再根据实验需求增加样品量。
注射用配方
注射用配方1: DMSO : Tween 80: Saline = 10 : 5 : 85 (如: 100 μL DMSO → 50 μL Tween 80 → 850 μL Saline)(IP/IV/IM/SC等) *生理盐水/Saline的制备:将0.9g氯化钠/NaCl溶解在100 mL ddH ₂ O中,得到澄清溶液。 注射用配方 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (如: 100 μL DMSO → 400 μL PEG300 → 50 μL Tween 80 → 450 μL Saline) 注射用配方 3: DMSO : Corn oil = 10 : 90 (如: 100 μL DMSO → 900 μL Corn oil) 示例: 以注射用配方 3 (DMSO : Corn oil = 10 : 90) 为例说明, 如果要配制 1 mL 2.5 mg/mL的工作液, 您可以取 100 μL 25 mg/mL 澄清的 DMSO 储备液,加到 900 μL Corn oil/玉米油中, 混合均匀。 View More
注射用配方 4: DMSO : 20% SBE-β-CD in Saline = 10 : 90 [如:100 μL DMSO → 900 μL (20% SBE-β-CD in Saline)] 口服配方
口服配方 1: 悬浮于0.5% CMC Na (羧甲基纤维素钠) 口服配方 2: 悬浮于0.5% Carboxymethyl cellulose (羧甲基纤维素) 示例: 以口服配方 1 (悬浮于 0.5% CMC Na)为例说明, 如果要配制 100 mL 2.5 mg/mL 的工作液, 您可以先取0.5g CMC Na并将其溶解于100mL ddH2O中,得到0.5%CMC-Na澄清溶液;然后将250 mg待测化合物加到100 mL前述 0.5%CMC Na溶液中,得到悬浮液。 View More
口服配方 3: 溶解于 PEG400 (聚乙二醇400) 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 0.7877 mL | 3.9387 mL | 7.8773 mL | |
| 5 mM | 0.1575 mL | 0.7877 mL | 1.5755 mL | |
| 10 mM | 0.0788 mL | 0.3939 mL | 0.7877 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1