
| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 5mg |
|
||
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| 500mg |
|
||
| Other Sizes |
|
| 靶点 |
HIV Reverse transcription (IC50 = 119 nM); NNRTI
|
|---|---|
| 体外研究 (In Vitro) |
与针对分离 RT 酶的有希望的结果一致,lerivirine 对多种耐药性和野生型 HIV 病毒株显示出出色的功效 [1]。
Lersivirine是第二代NNRTI,正在进行临床开发,用于治疗HIV-1。Lersivirine在结构上与依法韦仑不同,并以一种新的方式结合RT酶[2]。 |
| 体内研究 (In Vivo) |
Lersivirine 会导致交配的 Crl:CD1(ICR) 小鼠骨骼变异,这与发育迟缓和胎儿骨化减少有关。 Lersivirine(口服管饲;0、150、350 和 500 mg/kg;每天一次;妊娠第 6 至 17 天,随后在妊娠第 18 天进行剖腹产)在前 2 天以 250 mg 诱导肝代谢酶/ kg,然后将剂量增加至 500 mg/kg/天。
产妇和剖宫产观察[2] 治疗对产妇存活、流产或早产没有影响;21、19、18和22只雌性分别以0、150、350和500mg/kg/天的剂量产仔(表1)。Lersivirine/勒西韦林治疗不影响体重和体重增加;在150、350和500mg/kg/天的剂量下,GD 18的体重分别为对照组的100%、99%和102%(表2)。饲料消耗量在350和500mg/kg/天时减少(表2)。在350和500 mg/kg/天的剂量下,整个给药期(GD 6-18)的饲料消耗量明显低于同期对照组的值(p<0.05或p<0.01)(分别为对照组的92%和90%)。 胎儿形态学[2] 母体接受Lersivirine/雷西韦林治疗后,未出现任何外部、内脏或骨骼畸形(表4)。在350mg/kg/天和500mg/kg/天的三个胎儿(1.3%/窝)的外部检查中观察到腭裂内侧裂;然而,鉴于该实验室的背景发病率为每窝3.2%,因此认为该发病率与雷西韦林无关。在0、150、350和500mg/kg/天的剂量下,分别有5只(2.1%/窝)、4只(1.6%/窝),5只(2.1%/窝)和2只(0.7%/窝)出现前肢和/或后肢旋转;然而,鉴于发病率没有剂量反应性增加,这一发现不能归因于雷西韦林。500 mg/kg/天组中发生的唯一骨骼畸形是两个(1.4%/窝)胎儿的腰椎弓融合,鉴于缺乏其他骨骼畸形发生的证据,单窝两个胎儿的这种情况不是由雷西韦林引起的。 |
| 动物实验 |
Animals and Treatment [2]
One hundred presumed pregnant female Crl:CD1(ICR) mice were randomly assigned to four groups of 25 mice per group. Sixty-three additional presumed pregnant mice were assigned for use in toxicokinetic sample collection; control group of 9 and 18 per group for lersivirine dose groups. Mice were approximately 72 days of age and approximately 28 gm in weight upon arrival. Mice were individually housed in stainless steel, wire-bottomed cages. Water and feed were given ad libitum and the room was on a 12-hr light/dark cycle. Suspensions of lersivirine in 0.5% methylcellulose aqueous solution with 0.1% Tween 80 were administered orally via gavage once daily on days 6 through 17 of presumed gestation (gestation days 6–17) at doses of 0, 150, 350, and 500 mg/kg/day at a dosage volume of 10 ml/kg. At the highest dose tested (500 mg/kg/day), an initial dose of 250 mg/kg/day at a dosage volume of 5 ml/kg was administered for the first 2 days of dosage administration after which the mice were treated at 500 mg/kg/day. This dosing regimen allowed for maintenance of the maximum tolerable exposure following metabolizing enzyme induction; lersivirine has previously been shown to induce metabolism in rodents (Walker et al., 2009). The doses were based on a preliminary study in which pregnant mice (10/group) were dosed from GD 6 to 17 at doses of 150, 350, and 700 mg/kg/day (the 700 mg/kg/day group was dosed the first 2 days at 350 mg/kg/day). In this study, 4 of 10 mice in the 700 mg/kg/day group were euthanized moribund; all other animals survived until the scheduled euthanasia. Therefore, 500 mg/kg/day (with first 2 dose days at 250 mg/kg/day) was estimated to be the maximum tolerated dose in pregnant mice. Assessment of Lersivirine Exposure [2] On GD 17, blood samples (approximately 0.5 ml) were collected from the vena cava of three mice/group/time point after sacrifice. For lersivirine-treated mice, samples were collected before dosage and at approximately 0.5, 2, 4, 8, and 24 hr post-dose. Blood samples were transferred into lithium heparin–coated tubes, plasma was separated from whole blood by centrifugation and stored frozen at –20°C until analyzed. Plasma concentrations of lersivirine were determined by a validated HPLC assay. |
| 药代性质 (ADME/PK) |
Maternal Toxicokinetics [2]
Plasma levels of lersivirine on GD 17 following administration of lersivirine from GD 6 to 17 are presented in Table 6. Maternal plasma lersivirine exposure was similar across all three dose groups. |
| 毒性/毒理 (Toxicokinetics/TK) |
Lersivirine is a second-generation nonnucleoside reverse transcriptase inhibitor undergoing clinical development for the treatment of HIV-1. An embryo-fetal developmental toxicity study was performed to evaluate the maternal and developmental toxicity of lersivirine in pregnant mice. Mated Crl:CD1(ICR) mice were administered 0, 150, 350, and 500 mg/kg lersivirine once daily by oral gavage on gestation days 6 to 17, followed by cesarean section on gestation day 18. The first 2 days of dosing for the high-dose group were done at 250 mg/kg to allow induction of hepatic metabolizing enzymes, after which the dose was increased to 500 mg/kg/day. This dosing paradigm allowed for maintenance of exposure in the high-dose group despite the considerable autoinduction that occurs in rodents following lersivirine treatment. Lersivirine did not cause an increase in external, visceral, or skeletal malformations. Intrauterine growth retardation, demonstrated by reduced fetal body weights and increased variations associated with delayed skeletal ossification, was noted at 350 and 500 mg/kg/day. The results of these studies indicate that lersivirine is not teratogenic in mice.[2]
|
| 参考文献 | |
| 其他信息 |
Lersivirine is an aromatic ether.
Lersivirine has been used in trials studying the treatment of HIV-1. Lersivirine is a next-generation, pyrazole non-nucleoside reverse transcriptase inhibitor. Lersivirine retains activity against HIV viruses with mutations at position Y181, which confers resistance to efavirenz, etravirine, and nevirapine. The results of this study indicate that lersivirine is not teratogenic in mice. In addition, lersivirine was not teratogenic in rabbits (Campion et al., same issue). The results of the lersivirine studies, in concert with the apparent lack of signal for teratogenesis with other first-generation NNRTIs such as delavirdine and nevirapine (Watts et al., 2004), support a conclusion that the teratogenic signal attributed to efavirenz (Lewis-Hall, 2005; Mofenson, 2005), if real, is specific to that compound and does not represent an NNRTI class effect. While no fetal malformations were attributed to lersivirine treatment, developmental toxicity was noted in fetuses from mothers treated with lersivirine at 350 and 500 mg/kg/day. The primary expression of developmental toxicity was intrauterine growth retardation, demonstrated by reduced fetal weight, increased skeletal variations, and decreased ossification sites. The skeletal variations that were increased were mainly those associated with delayed ossification and interrupted ribs. Skeletal variations are defined as common findings that occur in the experimental species and strain that represent reversible delays or accelerations in development. This is in contrast to fetal malformations that are defined as irreversible changes that occur at low incidences in this species and strain. The primary measure of intrauterine growth retardation is fetal body weight. Fetal body weights were 16 and 21% less than controls at 350 and 500 mg/kg/day, respectively; with no effect on fetal body weight at 150 mg/kg/day. In addition to reduced fetal body weight, skeletal variations indicative of developmental delay that were attributed to maternal treatment with lersivirine at 350 and 500 mg/kg/day were incomplete ossification of the supraoccipital, nasals, frontals, cervical arches, pubes, or ischia, and absence of an ossified supraoccipital. In addition, reduced ossification was noted for the hyoid, caudal vertebrae, sternal centers, forelimb phalanges, hindlimb tarsals, and hindlimb phalanges in the 350 and 500 mg/kg/day groups. Ossification delays are well known to correlate with reductions in fetal weight in mice (Deol and Truslove, 1957; McLaren and Michie, 1958) that are considered unrelated to pathogenesis associated with structural malformations (Grüneberg, 1955). In addition to intrauterine growth retardation, two dead fetuses were found in the 500 mg/kg/day treatment group. There was no other evidence of impaired fetal viability, the numbers of viable fetuses were comparable between groups and lersivirine treatment was not associated with increases in resorbed fetuses. However, because the incidence of dead fetuses is historically low, a potential relationship to treatment could not be dismissed. Maternal plasma lersivirine exposure on GD 17 following repeated daily dosing from GD 6 was essentially flat across the approximately threefold increase in dose from 150 to 500 mg/kg/day. Lersivirine is characterized by moderate clearance and volume of distribution leading to good oral bioavailability and all of the clearance is hepatic (Allan et al., 2008). However, lersivirine administration to rodents causes hepatic enzyme induction and this autoinduction limits the systemic exposure that can be achieved at steady state (Walker et al., 2009). The extent of autoinduction results in a decrease in AUC (area under curve)(0–24 hr), exposure of 8- to 10-fold between day 1 and day 12 of dosing in mice (Walker et al., 2009). Therefore, while GD 17 exposures are essentially flat across dose groups, the day 1 exposure over the same dose range showed approximately 5- and approximately 10-fold increase in Cmax and AUC(0–24 hr), respectively (unpublished data from 1-month toxicology study). Therefore, although not measured in this study, lersivirine exposure over this dose range during the period of early organogenesis likely had a dose proportional increase not indicated by the GD 17 measurements, and would have showed toxicologically relevant differences in exposures between dose levels. In conclusion, the results of this study demonstrated that lersivirine treatment does not cause fetal malformations in mice. The primary effect produced by maternal lersivirine exposure was intrauterine growth retardation as indicated by reduced fetal weight, and skeletal variations associated with delays in ossification.[2] We prepared three discreet cohorts of potent non-nucleoside HIV reverse transcriptase inhibitors (NNRTIs) based on the recently reported 3-cyanophenoxypyrazole lead 3. Several of these compounds displayed very promising anti-HIV activity in vitro, safety, pharmacokinetic and pharmaceutical profiles. We describe our analysis and conclusions leading to the selection of alcohol 5 (UK-453,061, lersivirine) for clinical development.[1] |
| 分子式 |
C17H18N4O2
|
|---|---|
| 分子量 |
310.3504
|
| 精确质量 |
310.142
|
| 元素分析 |
C, 65.79; H, 5.85; N, 18.05; O, 10.31
|
| CAS号 |
473921-12-9
|
| 相关CAS号 |
147362-57-0 (Loviride); 16837-52-8 (Oxymatrine)
|
| PubChem CID |
16739244
|
| 外观&性状 |
Light yellow to khaki solid powder
|
| 密度 |
1.2±0.1 g/cm3
|
| 沸点 |
455.4±45.0 °C at 760 mmHg
|
| 闪点 |
229.2±28.7 °C
|
| 蒸汽压 |
0.0±1.2 mmHg at 25°C
|
| 折射率 |
1.595
|
| LogP |
3.3
|
| tPSA |
94.86
|
| 氢键供体(HBD)数目 |
1
|
| 氢键受体(HBA)数目 |
5
|
| 可旋转键数目(RBC) |
6
|
| 重原子数目 |
23
|
| 分子复杂度/Complexity |
456
|
| 定义原子立体中心数目 |
0
|
| InChi Key |
MCPUZZJBAHRIPO-UHFFFAOYSA-N
|
| InChi Code |
InChI=1S/C17H18N4O2/c1-3-15-17(16(4-2)21(20-15)5-6-22)23-14-8-12(10-18)7-13(9-14)11-19/h7-9,22H,3-6H2,1-2H3
|
| 化学名 |
5-((3,5-diethyl-1-(2-hydroxyethyl)-1H-pyrazol-4-yl)oxy)isophthalonitrile
|
| 别名 |
UK 453061; UK-453061; UK-453,061; UK453,061; Lersivirine; 473921-12-9; 3-CYANO-5-[[3,5-DIETHYL-1-(2-HYDROXYETHYL)-1H-PYRAZOL-4-YL]OXY]BENZONITRILE; UK-453,061; 5-((3,5-diethyl-1-(2-hydroxyethyl)-1H-pyrazol-4-yl)oxy)isophthalonitrile; 5-[3,5-diethyl-1-(2-hydroxyethyl)pyrazol-4-yl]oxybenzene-1,3-dicarbonitrile; R3ZGC15A9A; UK453061; UK 453,061
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
DMSO : ~50 mg/mL (~161.11 mM)
|
|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 3 mg/mL (9.67 mM) (饱和度未知) in 10% DMSO + 40% PEG300 +5% Tween-80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 30.0 mg/mL 澄清的 DMSO 储备液加入到400 μL PEG300中,混匀;再向上述溶液中加入50 μL Tween-80 +,混匀;然后加入450 μL 生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 3.2222 mL | 16.1108 mL | 32.2217 mL | |
| 5 mM | 0.6444 mL | 3.2222 mL | 6.4443 mL | |
| 10 mM | 0.3222 mL | 1.6111 mL | 3.2222 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
 NNRT-IN-5
NNRT-IN-5
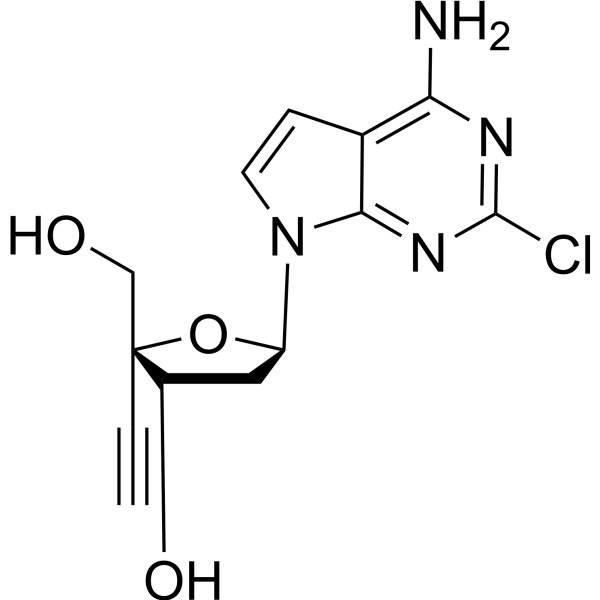 MK-8527
MK-8527
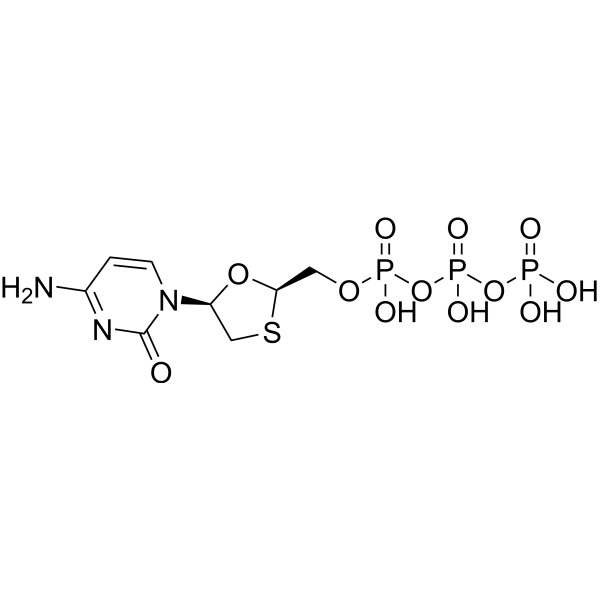 Lamivudine triphosphate
Lamivudine triphosphate
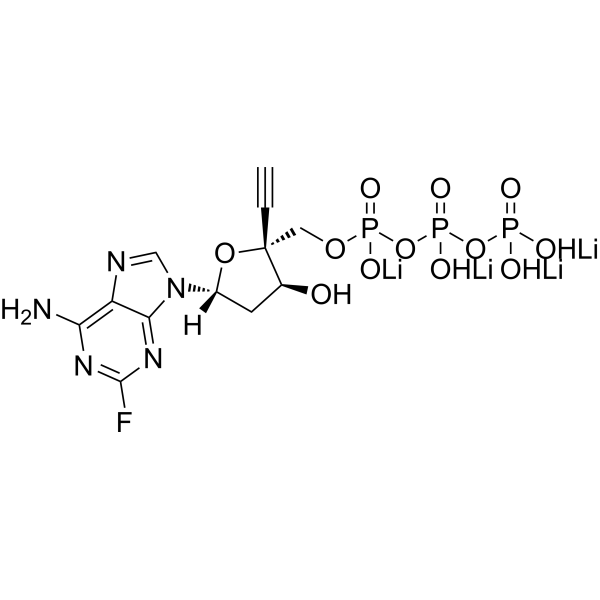 EFdA-TP tetralithium
EFdA-TP tetralithium
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1






























 COA
COA
