| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 1mg |
|
||
| 5mg |
|
||
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| Other Sizes |
|
| 靶点 |
EGFR(L858R/T790M) (IC50 = 0.26 nM)
|
|---|---|
| 体外研究 (In Vitro) |
JBJ-04-125-02对EGFRL858R/T790M的IC50为0.26 nM,是一种具有突变结构和EGFR活性的强EGFR抑制剂[1]。
|
| 体内研究 (In Vivo) |
基于体外单药活性,我们试图确定JBJ-04-125-02在体内是否也有效。JBJ-04-125-02静脉注射3 mg/kg剂量后,半衰期适中,为3小时,曲线下面积较高,为728577min·ng/mL(AUClast)。口服20mg/kg剂量的JBJ-04-125-02,平均最大血药浓度为1.1μM,口服生物利用度仅为3%(表S3A)。基于这些发现,我们进行了一项药效学研究,其中EGFR L858R/T790M/C797S基因工程小鼠(GEM)在肿瘤发生后,每天口服一次,用3剂赋形剂或100mg/kg JBJ-02-112-05或50mg/kg或100mg/kg的JBJ-04-125-02治疗,并评估其对EGFR磷酸化和下游信号传导的影响(图2C)。50mg/kg和100mg/kg剂量均有效抑制了EGFR、AKT和ERK1/2的磷酸化(图2C)。JBJ-02-112-05(100mg/kg)也抑制了EGFR和下游信号通路的磷酸化,尽管不如JBJ-04-125-02(图2C)强烈。在随后的疗效研究中,我们用赋形剂、100mg/kg的JBJ-02-112-05或50mg/kg的JBJ-04-125-02治疗EGFR L858R/T790M/C797S GEM小鼠,并通过连续MRI成像跟踪肿瘤体积的变化。尽管JBJ-02-112-05(表S3A)的药代动力学特征优于JBJ-04-125-02,但在疗效研究中无效,肿瘤生长与赋形剂对照相似。相比之下,JBJ-04-125-02治疗在治疗4周内导致明显的肿瘤消退(图2D),并持续治疗15周(图2E)。尽管JBJ-04-125-02的口服生物利用度较差,但长期治疗导致药物在血浆和肿瘤中积聚,这可能是其疗效的原因(表S3B)。值得注意的是,JBJ-04-125-02治疗与体重减轻或明显的毒性症状无关(图S2A和数据未显示)[1]。
|
| 酶活实验 |
基于HTRF的EGFR生化检测[1]
如前所述,在哈佛医学院ICCB Longwood筛查设施,使用均相时间分辨荧光(HTRF)KinEASE TK测定法进行了L858R/T790M EGFR的生化测定。酶浓度为20pM,ATP浓度为100µM。使用D300数字分配器将DMSO中的抑制剂化合物直接分配到384孔板中,然后立即使用多点组合试剂分配器添加缓冲水溶液。IC50值用11或23点抑制曲线测定。 EGFR蛋白表达和纯化[1] 使用pTriEX系统以His6和GST融合双标记形式制备跨越人EGFR残基696-1022的构建体(包括野生型、L858R/T790M、L858R-T790M/C797S和T790M/V948R突变序列),用于在Sf9昆虫细胞中表达,基本如上所述。通过Ni-NTA和谷胱甘肽亲和层析纯化EGFR激酶蛋白,然后按照既定程序用TEV切割后进行尺寸排阻层析,以去除His6-GST融合伴侣。 |
| 细胞实验 |
细胞活力测定[1]
Ba/F3、H1975、H3255GR和H3255DR细胞用越来越高浓度的抑制剂处理72小时,并根据先前建立的方法通过MTS测定评估生长或生长抑制。对于研究JBJ-04-125-02在EGF或西妥昔单抗存在下的作用的实验,在用抑制剂处理细胞的同时加入10ng/ml的EGF、1μg/ml或10μg/ml的西妥昔珠单抗。 生物素化药物下拉试验[1] 对于生化下拉分析,将10g游离EGFR-L858R/T790M或奥西咪替尼标记的EGFR-L85R/T790M与链霉抗生物素蛋白偶联的琼脂糖珠和4倍摩尔过量的生物素化JBJ-04-125-02化合物在4°C下孵育一小时。通过离心回收链霉抗生物素蛋白珠,用10倍床体积的TBS缓冲液洗涤三次,并通过SDS-PAGE分析结合的蛋白质。奥西美替尼标记的EGFR-L858R/T790M是通过在奥西美替尼存在下纯化EGFR-L858R/T790M制备的;将100μM奥西咪替尼加入昆虫细胞裂解物中,在最终尺寸排除步骤之前,将2μM奥西咪替尼保持在纯化缓冲液中。质谱法证实了奥西替尼的化学计量标记 对于体外下拉试验,细胞用剂量递增的WZ-4002、奥西咪替尼或阿法替尼处理两小时,然后进行裂解和蛋白质定量。将500g蛋白质与生物素化接头(对照)或生物素化JBJ-04-125-02一起孵育两小时,然后加入50%NeutrAvidin琼脂糖珠浆一小时,在抑制剂剂量增加的情况下沉淀与生物素基化变构抑制剂相关的EGFR。用含有1%IGEPAL的PBS洗涤珠粒,去除多余的缓冲液,然后将样品悬浮在2X SDS样品制备缓冲液中进行蛋白质印迹分析。 交联分析[1] H1975和H3255GR细胞用冰冷的PBS洗涤两次,然后用1mM BS3在PBS中孵育30分钟,然后用20mM Tris-HCl pH 7.4淬灭反应15分钟。然后用冰冷的PBS再次洗涤细胞两次,然后在NP40裂解缓冲液中裂解细胞并进行蛋白质印迹分析。 |
| 动物实验 |
Pharmacokinetic studies[1]
All procedures described are covered under existing protocols and have been approved by the Scripps Florida IACUC to be conducted in the Scripps vivarium, which is fully AAALAC accredited. Pharmacokinetics were determined in n=3 male C57Bl/6 mice. Compounds were dosed as indicated in the text via intravenous tail vein injection or by oral gavage. Blood was collected using minimal sampling techniques where ~25 µL blood is collected from a small nick in the tail using Li-heparin-coated hematocrit tubes at 5min, 15min, 30min, 1h, 2h, 4h, 6h, and 8h. Plasma was generated via centrifugation using a hematocrit rotor. Plasma concentration was determined via LC-MS/MS by comparison of the analyte/IS peak area using a nine-point standard curve between 0.4ng/mL and 2000ng/mL prepared in mouse plasma. Pharmacokinetic analysis was done with WinNonlin, Centara inc. using a noncompartmental model. In vivo studies[1] The EGFR L858R/T790M/C797S mutant mouse cohort was established and reported previously. These mice were monitored by MRI to quantify lung tumor burden before being assigned to various treatment study cohorts. All the treatment mice had equal initial tumor burden before starting treatment. H1975 and DFCI282 cells were grown subcutaneously in Nu/Nu mice purchased from Charles River Laboratories International Inc. Mice were randomly grouped, and treatment initiated when tumor size reached 100 to 200 mm3. Each cohort included at least 5 mice. For pharmacodynamic (PD) studies, tumors were harvested at 3 hours after the last dose. For single agent efficacy studies, GEM and H1975 xenograft mice were treated for up to 15 weeks daily and 30 days respectively and monitored daily. For combination efficacy studies, H1975 and DFCI282 xenograft mice were treated for 28 days. Drugs were then withdrawn and mice were monitored daily for up to 101 days and 31 days respectively. Tumor size of all mice were monitored, and volumes were calculated using the following formula: (mm3) = length × width × width × 0.5. All mice were sacrificed when tumor volume reached approximately 2000mm3. [1] Both JBJ-02-112-05 and JBJ-04-125-02 were dissolved in 5% NMP (5% 1-methyl-2-pyrrolidinone: 95% PEG-300). Osimertinib was dissolved in 0.5% HMPC (0.5% Hydroxypropyl methylcellulose: 99.5% 0.05N hydrogen chloride). Mice received 2.5mg/kg or 25mg/kg osimertinib once daily orally. |
| 参考文献 | |
| 其他信息 |
Allosteric kinase inhibitors offer a potentially complementary therapeutic strategy to ATP-competitive kinase inhibitors due to their distinct sites of target binding. In this study, we identify and study a mutant-selective EGFR allosteric inhibitor, JBJ-04-125-02, which as a single agent can inhibit cell proliferation and EGFRL858R/T790M/C797S signaling in vitro and in vivo. However, increased EGFR dimer formation limits treatment efficacy and leads to drug resistance. Remarkably, osimertinib, an ATP-competitive covalent EGFR inhibitor, uniquely and significantly enhances the binding of JBJ-04-125-02 for mutant EGFR. The combination of osimertinib and JBJ-04-125-02 results in an increase in apoptosis, a more effective inhibition of cellular growth, and an increased efficacy in vitro and in vivo compared with either single agent alone. Collectively, our findings suggest that the combination of a covalent mutant-selective ATP-competitive inhibitor and an allosteric EGFR inhibitor may be an effective therapeutic approach for patients with EGFR-mutant lung cancer. SIGNIFICANCE: The clinical efficacy of EGFR tyrosine kinase inhibitors (TKI) in EGFR-mutant lung cancer is limited by acquired drug resistance, thus highlighting the need for alternative strategies to inhibit EGFR. Here, we identify a mutant EGFR allosteric inhibitor that is effective as a single agent and in combination with the EGFR TKI osimertinib.[1]
|
| 分子式 |
C29H26FN5O3S
|
|---|---|
| 分子量 |
543.61184835434
|
| 精确质量 |
543.17
|
| 元素分析 |
C, 64.07; H, 4.82; F, 3.49; N, 12.88; O, 8.83; S, 5.90
|
| CAS号 |
2140807-05-0
|
| 相关CAS号 |
JBJ-04-125-02;2060610-53-7; 2140807-05-0 (racemic) ; 2060610-53-7 (R-isomer)
|
| PubChem CID |
132020316
|
| 外观&性状 |
White to off-white solid powder
|
| LogP |
3.8
|
| tPSA |
126Ų
|
| 氢键供体(HBD)数目 |
3
|
| 氢键受体(HBA)数目 |
8
|
| 可旋转键数目(RBC) |
6
|
| 重原子数目 |
39
|
| 分子复杂度/Complexity |
870
|
| 定义原子立体中心数目 |
0
|
| InChi Key |
VHQVOTINPRYDAO-UHFFFAOYSA-N
|
| InChi Code |
InChI=1S/C29H26FN5O3S/c30-21-5-8-25(36)24(16-21)26(27(37)33-29-32-11-14-39-29)35-17-20-2-1-19(15-23(20)28(35)38)18-3-6-22(7-4-18)34-12-9-31-10-13-34/h1-8,11,14-16,26,31,36H,9-10,12-13,17H2,(H,32,33,37)
|
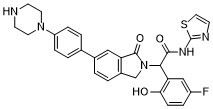
| 化学名 |
2-(5-fluoro-2-hydroxyphenyl)-2-[3-oxo-5-(4-piperazin-1-ylphenyl)-1H-isoindol-2-yl]-N-(1,3-thiazol-2-yl)acetamide
|
| 别名 |
JBJ-04-125-02 racemate; JBJ0412502; JBJ-04-125-02; JBJ04-125-02; JBJ 0412502; JBJ 04-125-02; JBJ-0412502
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
DMSO: 100~250 mg/mL (184.0~459.9 mM)
|
|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: 2.08 mg/mL (3.83 mM) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 悬浮液;超声助溶。
例如,若需制备1 mL的工作液,可将100 μL 20.8 mg/mL澄清DMSO储备液加入400 μL PEG300中,混匀;然后向上述溶液中加入50 μL Tween-80,混匀;加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: 2.08 mg/mL (3.83 mM) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 悬浊液; 超声助溶。 例如,若需制备1 mL的工作液,可将 100 μL 20.8 mg/mL澄清DMSO储备液加入900 μL 20% SBE-β-CD生理盐水溶液中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 View More
配方 3 中的溶解度: ≥ 2.08 mg/mL (3.83 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.8396 mL | 9.1978 mL | 18.3955 mL | |
| 5 mM | 0.3679 mL | 1.8396 mL | 3.6791 mL | |
| 10 mM | 0.1840 mL | 0.9198 mL | 1.8396 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
|
|
|
|
|
 PD153035 HCl (SU5271; ZM252868)
PD153035 HCl (SU5271; ZM252868)
 RG 13022
RG 13022
 AV-412
AV-412
 O-去甲基埃罗替尼盐酸盐
O-去甲基埃罗替尼盐酸盐
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1



































