
| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 10 mM * 1 mL in DMSO |
|
||
| 1mg |
|
||
| 5mg |
|
||
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| Other Sizes |
|
| 靶点 |
IDH1R132H/isocitrate dehydrogenase 1 mutant (12 nM)
|
|---|---|
| 体外研究 (In Vitro) |
Ivosidenib (AG-120)(0-13 μM;48 小时)以相当的效力和 IC50 值抑制许多 IDH1-R132 突变体:IDH1-R132H (IC50=12 nM); IDH1-R132C (IC50=13 nM); IDH1-R132G (IC50=8 nM); IDH1-R132L (IC50=13 nM);和 IDH1-R132S (IC50=12 nM),分别 [1]。
生化和细胞生物学分析显示,Ivosidenib/AG-120抑制了几种IDH1-R132突变体,其效力与R132H相似(表3),对IDH1亚型具有高度选择性,在微摩尔浓度下对IDH2(WT或突变体)亚型没有抑制作用(表S2)。100μM的AG-120没有抑制所测试的多种脱氢酶(表S3)。[1] 在体外,AG-120对mIDH1-R132同二聚体表现出快速的平衡抑制作用。由于所有可溶性mIDH1酶制剂中都存在持续的预结合NADP(H),因此结合动力学研究以证明作用模式尚无定论(支持信息,图S1和S2)。令人惊讶的是,Ivosidenib/AG-120对IDH1-WT同二聚体表现出缓慢的紧密结合抑制作用(图S3和S4)。[1] AG-120/Ivosidenib在多种mIDH1-R132内源性和过表达细胞系中也显示出良好的细胞效力(表3),表明其可用于所有mIDH1-R16癌症。[1] IDH突变已被证明可以通过表观遗传和代谢重连阻断正常的细胞分化。1,3−5为了确定mIDH1抑制对原代人类AML母细胞的影响,在离体试验中用Ivosidenib/AG-120处理患者的骨髓或外周血样本(表S5),mIDH1-R132H、mIDH1-R232C和IDH1-WT。在AG-120存在或不存在的情况下,在含有细胞因子(密度为0.5×106个细胞/mL)的培养基中分选和培养活母细胞。在mIDH1样本中,AG-120在最低测试剂量(0.5μM)下将细胞内2-HG水平降低了96%,在1和5μM下分别降低了98.6%和99.7%(图2)。在评估的多个IDH1-WT患者样本中无法测量2-HG。AG-120诱导AML患者离体治疗的原代mIDH1-R132H和mIDH1-R232C(但不是IDH1-WT)母细胞分化,如甲基纤维素测定中形成分化集落的能力增强、细胞表面分化标志物水平升高以及成熟髓系细胞比例增加所示[1]。 |
| 体内研究 (In Vivo) |
治疗后 12 小时,AG-120(灌胃给药;50 mg/kg 和 150 mg/kg)产生最大抑制(50 mg/kg 和 150 mg/kg 剂量分别为 92.0% 和 95.2%)并迅速减少肿瘤2-HG 浓度 [1]。
在Sprague-Dawley大鼠、比格犬和食蟹猴中进行的IvosidenibPK研究显示,口服吸收迅速,全身血浆清除率(CLp)低,半衰期中长期(t1/2)(表S4)。尽管在啮齿类动物的重复研究中观察到中度暴露减少(数据未显示),但食蟹猴的暴露没有减少,癌症患者在多次给药后观察到长时间的t1/2和Ivosidenib/AG-120的积累。 对血脑屏障完整的大鼠单次口服50mg/kg剂量后,Ivosidenib/AG-120的脑穿透率为4.1%(AUC0-8h[脑]/AUC0-8h[血浆])。然而,血脑屏障受损的神经胶质瘤患者的脑穿透率可能更高。鉴于AG-120非常有效且耐受性良好,它有可能在大脑中达到治疗浓度,其对胶质瘤的治疗益处正在临床试验中进行评估。 AG-120/Ivosidenib在接种HT1080细胞的雌性裸BALB/c小鼠中显示出明显的肿瘤2-HG减少。每只小鼠通过强饲法接受50或150mg/kg的单次口服赋形剂或AG-120。肿瘤2-HG浓度迅速下降,在给药后约12小时达到最大抑制率(50mg/kg和150mg/kg剂量分别为92.0%和95.2%)。单剂量AG-120后48-72小时,肿瘤2-HG浓度接近基线水平(图1),这与AG-120抑制的可逆性一致[1]。 |
| 酶活实验 |
使用黄递酶/雷沙祖林偶联系统测定化合物对mIDH1-R132H酶反应的抑制效力[1]
在初级反应中,α-KG酸还原为D-2-羟基戊二酸(2-HG)伴随着NADPH氧化为NADP。在二次黄递酶/雷沙祖林反应中测量反应时间结束时残留的NADPH的量,其中NADPH以1:1的摩尔比消耗,雷沙祖林转化为高荧光的间苯二酚。未抑制的反应在测定结束时显示出低荧光,而mIDH1-R132H对NADPH的消耗已被小分子抑制的反应显示出高荧光。初级反应在50的体积中进行L 1X缓冲液(150 mM NaCl,20 mM Tris 7.5,10 mM MgCl2,0.05%w/v牛血清白蛋白[BSA]),含有2 nM mIDH1-R132H,1 mMα-KG和4M NADPH,并在25°C下进行60分钟。为了进行二次反应,25包含36的1X缓冲区的Lg/mL的黄递酶和30mM的10-雷沙祖林加入到主要反应中,并在25°C下再孵育10分钟。在Spectramax平板阅读器上以Ex 544 Em 590读取Florence。如前所述表达并纯化重组蛋白。5以100%二甲基亚砜(DMSO)浓度制备化合物或化合物稀释液,并以1:100稀释到最终反应中。mIDH1-R132C在类似条件下进行测定,不同之处在于1X缓冲液为50mM K2HPO4 pH 6.5、40mM NaHCO3、5mM MgCl2、10%甘油、0.03%w/v BSA。 IDH1-WT酶反应测定抑制剂效力[1] IDH1-WT酶是在mIDH1-R132H测定法的改良版中测定的。由于该酶将NADP转化为NADPH,异柠檬酸转化为α-KG,因此NADPH产物可以通过直接偶联到黄递酶/重氮祖林系统并读取Ex 544 Em 590下的再吸收蛋白产量来连续测定。在50个样本中进行了分析L 1X缓冲液(150 mM NaCl,20 mM Tris pH 7.5,10 mM MgCl2,0.05%(w/v)BSA,2 mMβ-巯基乙醇[B-ME]),含有50M NADP,70M DL异柠檬酸盐和31.2ng/mL IDH1-WT酶(反应时间1或16小时)。直接耦合系统包括20g/mL黄递酶和40M resazurin. IDH2-WT酶反应测定抑制剂效力[1] 化合物对IDH2-WT酶的抑制效力在与黄递酶的偶联测定中测定。在该测定中,IDH2-WT产生NADPH与雷萨祖林同时还原为高荧光的间苯二酚有关。酶稀释至0.06g/mL,40L 1X测定缓冲液(150 mM NaCl,50 mM磷酸钾pH 7,10 mM MgCl2,10%甘油,2 mM B-ME,0.03%BSA),其中1L的化合物加入DMSO中。将混合物在室温(RT)下孵育16小时。反应开始时加入10基质混合物的L(200M异柠檬酸盐,175M NADP,60g/mL黄递酶,200M resazurin,在1X测定缓冲液中),并在RT下运行30分钟。加入25L的6%十二烷基硫酸钠,并在Spectramax平板阅读器上以Ex544/Em590读取 mIDH2-R140Q和mIDH2-R172K酶反应测定抑制剂效力[1] 在终点测定中测定了对mIDH2-R140Q和mIDH2-R172K酶的抑制效力,其中通过添加大量过量的黄递酶和雷沙唑林来测量反应结束时残留的NADPH的量。将mIDH2/R140Q 11稀释至0.25g/mL,40L 1X测定缓冲液(150 mM NaCl,50 mM磷酸钾,pH 7.5,10 mM MgCl2,10%甘油,2 mM B-ME,0.03%BSA),在25°C,1L二甲基亚砜中的化合物。反应开始时加入10基质混合物的L(20M NADPH,8Mα-KG,在1X测定缓冲液中),并在25°C下孵育1小时。然后,通过添加25L检测混合物(36g/mL黄递酶,18M resazurin在1X测定缓冲液中),在25°C下孵育5分钟,并如上所述读取。mIDH2-R172K的测定与mIDH2-R140Q的测定一样,具有以下修饰:1.25使用g/mL蛋白质,底物混合物含有50M NADPH和6.4Mα-KG,并且在开始反应之前将化合物孵育1小时。 |
| 细胞实验 |
将细胞以5000(U87MG、HCCC9810、COR-L 105)或2500(HT1080)个细胞/孔的密度接种在各自的生长培养基中至96孔微量滴定板中,并在37°C和5%CO2下孵育过夜。第二天,AG-120在100%DMSO中制备为10mM的原液,然后在培养基中稀释至0.1%DMSO的最终浓度。最高浓度剂量为3µM。从细胞板中取出培养基,并向每个孔中加入200µL的化合物稀释液。对于神经球,将化合物和细胞(40000/孔)同时接种在一起。在37°C下与化合物孵育48小时后,从每个孔中取出100µL培养基,并按如下所述进行分析。然后使细胞板再孵育24小时。在添加化合物后72小时,将10mL/板的Promega Cell Titer Glo试剂解冻并混合。将细胞板从培养箱中取出,并使其平衡至RT。然后向每孔培养基中加入100µL试剂。将细胞板置于轨道振荡器上10分钟,然后在RT下放置20分钟。然后以500ms的积分时间读取板的发光,以确定任何化合物对生长抑制的影响(细胞增殖的半最大抑制,GI50)。[1]
|
| 动物实验 |
Animal/Disease Models: Female BALB/c nude mice were inoculated with HT1080 cells [1]
Doses: 50 mg/kg and 150 mg/kg Route of Administration: intragastric (po) (po)administration; 50 mg/kg and 150 mg/kg Experimental Results: Mouse tumors were shown 2-HG was Dramatically diminished. Generation of HT1080 mIDH1-R132C xenografts[1] All animal studies were approved by the Institutional Animal Care and Use Committee and conducted in compliance with all national and local guidelines and regulations. HT1080 mIDH1-R132C cells were grown and 3 × 106 cells were inoculated subcutaneously on the flank of female BALB/c mice. When tumors reached approximately 200 mm3 the mice were randomized into dosing groups according to tumor size and treated with AG-120. Mice were dosed orally by gavage with a single dose of AG-120 at 50 or 150 mg/kg (n = 21 per dose group). Blood and tumor tissue samples were collected at 1, 3, 6, 12, 24, 48, and 72 hours following the dose (n = 3 at each time point) and were analyzed for AG-120 and 2-HG via LC-MS/MS |
| 药代性质 (ADME/PK) |
Absorption, Distribution and Excretion
Following oral administration, ivosidenib is rapidly absorbed. The Cmax following a single oral dose is 4503 ng/mL in patients with relapsed or refractory AML, 4820 ng/mL in patients with newly diagnosed AML who were also treated with azacitidine, and 4060 ng/mL in patients with cholangiocarcinoma. The steady-state was reached within 14 days. The steady-state Cmax is 6551 ng/mL in patients with relapsed or refractory AML, 6145 ng/mL in patients with newly diagnosed AML who were also treated with azacitidine, and 4799 ng/mL in patients with cholangiocarcinoma. The Tmax ranges from two to three hours. A high-fat meal increases ivosidenib exposure. Following oral administration of ivosidenib, about 77% of the dose was eliminated in feces, where 67% was in the form of unchanged parent drug. About 17% of the dose was excreted in urine, where 10% was in the form of unchanged ivosidenib. The apparent volume of distribution at steady state is 403 L in patients with relapsed or refractory AML, 504 L in patients with newly diagnosed AML who were also treated with azacitidine, and 706 L in patients with cholangiocarcinoma. The apparent clearance at steady state is 5.6 L/h in patients with relapsed or refractory AML, 4.6 L/h in patients with newly diagnosed AML who were also treated with azacitidine, and 6.1 L/h in patients with cholangiocarcinoma. Metabolism / Metabolites Ivosidenib is predominantly metabolized by CYP3A4 via oxidation. The exact chemical structures of the metabolites formed from CYP3A4-mediated oxidation have not been fully characterized. Ivosidenib can also undergo N-dealkylation and hydrolysis as minor metabolic pathways. Biological Half-Life The terminal half-life at steady state is 58 hours in patients with relapsed or refractory AML, 98 hours in patients with newly diagnosed AML who were also treated with azacitidine, and 129 hours in patients with cholangiocarcinoma. |
| 毒性/毒理 (Toxicokinetics/TK) |
Hepatotoxicity
Elevations in serum aminotransferase levels are common during ivosidenib therapy occurring in 15% to 20% of patients but rising above 5 times the upper limit of the normal range in only 1% to 2%. Ivosidenib has had limited clinical use but has not been linked to instances of acute liver injury with symptoms or jaundice. Because of the limited clinical experience with the use of IDH inhibitors, their potential for causing liver injury is not well defined. In prelicensure studies, ivosidenib therapy was associated with "differentiation syndrome" in 5% to 20% of patients, which was sometimes severe and life-threatening. Differentiation syndrome is marked by rapid proliferation of myeloid cells and symptoms of respiratory distress, accompanied by hypoxia, pulmonary infiltrates and pleural effusions. Other manifestations include renal impairment, fever, lymphadenopathy, bone pain, peripheral edema and weight gain. Liver dysfunction can also occur but is generally overshadowed by the more severe systemic manifestations. The onset of differentiation syndrome is generally within 2 to 8 weeks of starting therapy and the course can be severe. Management includes stopping ivosidenib and use of corticosteroids and hydroxyurea in more severe cases. Patients can be restarted on ivosidenib once the syndrome resolves. Likelihood score: E* (unproven but suspected cause of clinically apparent liver injury). Effects During Pregnancy and Lactation ◉ Summary of Use during Lactation No information is available on the clinical use of ivosidenib during breastfeeding. Because ivosidenib is 92 to 96% bound to plasma proteins, the amount in milk is likely to be low. However, its half-life is about 93 hours and it might accumulate in the infant. The manufacturer recommends that breastfeeding be discontinued during ivosidenib therapy and for 1 month after the dose. ◉ Effects in Breastfed Infants Relevant published information was not found as of the revision date. ◉ Effects on Lactation and Breastmilk Relevant published information was not found as of the revision date. Protein Binding _In vitro_, ivosidenib is 92-96% bound to plasma proteins. |
| 参考文献 | |
| 其他信息 |
Ivosidenib is a tertiary carboxamide resulting from the formal condensation of the carboxy group of (2S)-1-(4-cyanopyridin-2-yl)-5-oxopyrrolidine-2-carboxylic acid with the secondary amino group of (2S)-2-(2-chlorophenyl)-N-(3,3-difluorocyclobutyl)-2-[(5-fluoropyridin-3-yl)amino]acetamide. It is approved by the FDA for the treatment of acute myeloid leukemia (AML) in patients with an isocitrate dehydrogenase-1 (IDH1) mutation. It has a role as an antineoplastic agent and an EC 1.1.1.42 (isocitrate dehydrogenase) inhibitor. It is a member of monochlorobenzenes, a cyanopyridine, a member of pyrrolidin-2-ones, an organofluorine compound, a tertiary carboxamide and a secondary carboxamide.
Ivosidenib is a first-in-class isocitrate dehydrogenase-1 (IDH1) inhibitor. IDH1 is an enzyme that is often mutated and overexpressed in some cancers, leading to aberrant cell growth and proliferation. Ivosidenib inhibits mutated IDH1, blocking the enzymatic activity and further differentiation of cancer cells. Ivosidenib was granted accelerated approval by the FDA in July 2018 for the treatment of relapsed of refractory acute myeloid leukemia in adults. It is currently approved to also treat newly diagnosed acute myeloid leukemia in older adults in combination [azacitidine] or as monotherapy, as well as locally advanced or metastatic cholangiocarcinoma and relapsed or refractory myelodysplastic syndromes in adults. The drug is only effective in patients with a susceptible IDH1 mutation. In February 2023, the EMA's Committee for Medicinal Products for Human Use (CHMP) adopted a positive opinion of ivosidenib and recommended it be granted marketing authorization for the treatment of acute myeloid leukemia and cholangiocarcinoma. It was fully approved by the EMA in May 2023. Ivosidenib is an Isocitrate Dehydrogenase 1 Inhibitor. The mechanism of action of ivosidenib is as an Isocitrate Dehydrogenase 1 Inhibitor, and Cytochrome P450 3A4 Inducer, and Cytochrome P450 2C9 Inducer. Ivosidenib is an orally available small molecule inhibitor of mutated isocitrate dehydrogenase-1 that is used as an antineoplastic agent in the treatment of adults of acute myelogenous leukemia (AML). Ivosidenib is associated with a moderate rate of serum aminotransferase elevations during therapy and is suspected to be the cause of rare instances of clinically apparent acute liver injury. Ivosidenib is an orally available inhibitor of isocitrate dehydrogenase type 1 (IDH1), with potential antineoplastic activity. Upon administration, AG-120 specifically inhibits a mutated form of IDH1 in the cytoplasm, which inhibits the formation of the oncometabolite, 2-hydroxyglutarate (2HG). This may lead to both an induction of cellular differentiation and an inhibition of cellular proliferation in IDH1-expressing tumor cells. IDH1, an enzyme in the citric acid cycle, is mutated in a variety of cancers; it initiates and drives cancer growth by both blocking cell differentiation and catalyzing the formation of 2HG. IVOSIDENIB is a small molecule drug with a maximum clinical trial phase of IV (across all indications) that was first approved in 2018 and has 3 approved and 7 investigational indications. This drug has a black box warning from the FDA. Pharmacodynamics Ivosidenib is an antineoplastic agent that is effective in cancers with a susceptible IDH1 mutation, which indicates increased levels of oncometabolite D-2-hydroxyglutarate (D-2HG) in cancer cells. Ivosidenib decreases D-2HG levels in a dose-dependent manner by inhibiting the IDH1 enzyme. Ivosidenib inhibits both the mutant and wild-type IDH1 but does not inhibit IDH2. Somatic point mutations at a key arginine residue (R132) within the active site of the metabolic enzyme isocitrate dehydrogenase 1 (IDH1) confer a novel gain of function in cancer cells, resulting in the production of d-2-hydroxyglutarate (2-HG), an oncometabolite. Elevated 2-HG levels are implicated in epigenetic alterations and impaired cellular differentiation. IDH1 mutations have been described in an array of hematologic malignancies and solid tumors. Here, we report the discovery of AG-120 (ivosidenib), an inhibitor of the IDH1 mutant enzyme that exhibits profound 2-HG lowering in tumor models and the ability to effect differentiation of primary patient AML samples ex vivo. Preliminary data from phase 1 clinical trials enrolling patients with cancers harboring an IDH1 mutation indicate that AG-120 has an acceptable safety profile and clinical activity. Together, these compelling preclinical data provided the rationale to advance AG-120 into clinical development. The discovery of enasidenib, which is active against mIDH2, and now AG-120 (ivosidenib) against mIDH1 as described here, presents a novel class of cancer therapy based on cellular differentiation. AG-120 is a potent mIDH1 inhibitor with favorable nonclinical and clinical safety profiles that has shown promising clinical activity in phase 1 clinical trials for both solid and hematologic malignancies. In patients with relapsed/refractory mIDH1 AML, interim results from the ongoing phase 1 trial have demonstrated an overall response rate of 42% and a complete response rate of 22% (median duration of complete response 9.3 months).15 Long-term stable disease has been observed in patients with previously treated nonenhancing mIDH1 gliomas,16 and in heavily pretreated patients with mIDH1 cholangiocarcinoma, where the median progression-free survival was 3.8 months and the 6-month progression-free survival rate was 40%.17 In these two single arm, phase 1 studies, AG-120 has demonstrated an acceptable safety profile to date.15−18 AG-120 is currently in late-stage clinical development in adults with mIDH1 AML (ClinicalTrials.gov NCT03173248), and with previously treated advanced mIDH1 cholangiocarcinoma (NCT02989857).[1] |
| 分子式 |
C28H22CLF3N6O3
|
|---|---|
| 分子量 |
582.97
|
| 精确质量 |
582.139
|
| 元素分析 |
C, 57.69; H, 3.80; Cl, 6.08; F, 9.78; N, 14.42; O, 8.23
|
| CAS号 |
1448347-49-6
|
| 相关CAS号 |
(R,S)-Ivosidenib;2070009-31-1;IDH1 Inhibitor 8;1448346-63-1 (racemate)
|
| PubChem CID |
71657455
|
| 外观&性状 |
White to off-white solid
|
| 密度 |
1.5±0.1 g/cm3
|
| 沸点 |
854.3±65.0 °C at 760 mmHg
|
| 闪点 |
470.4±34.3 °C
|
| 蒸汽压 |
0.0±3.2 mmHg at 25°C
|
| 折射率 |
1.651
|
| LogP |
0.38
|
| tPSA |
119
|
| 氢键供体(HBD)数目 |
1
|
| 氢键受体(HBA)数目 |
9
|
| 可旋转键数目(RBC) |
7
|
| 重原子数目 |
41
|
| 分子复杂度/Complexity |
1050
|
| 定义原子立体中心数目 |
2
|
| SMILES |
ClC1=CC=CC=C1[C@@H](C(NC1CC(C1)(F)F)=O)N(C1C=NC=C(C=1)F)C([C@@H]1CCC(N1C1C=C(C#N)C=CN=1)=O)=O
|
| InChi Key |
WIJZXSAJMHAVGX-DHLKQENFSA-N
|
| InChi Code |
InChI=1S/C28H22ClF3N6O3/c29-21-4-2-1-3-20(21)25(26(40)36-18-11-28(31,32)12-18)37(19-10-17(30)14-34-15-19)27(41)22-5-6-24(39)38(22)23-9-16(13-33)7-8-35-23/h1-4,7-10,14-15,18,22,25H,5-6,11-12H2,(H,36,40)/t22-,25-/m0/s1
|
| 化学名 |
(S)-N-((S)-1-(2-chlorophenyl)-2-((3,3-difluorocyclobutyl)amino)-2-oxoethyl)-1-(4-cyanopyridin-2-yl)-N-(5-fluoropyridin-3-yl)-5-oxopyrrolidine-2-carboxamide
|
| 别名 |
RG-120; AG120; 1448347-49-6; Tibsovo; Q2PCN8MAM6; Ivosidenib [INN]; Ivosidenib [USAN]; trade name: Tibsovo®, AG-120; AG 120; RG 120; RG120
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
|
|||
|---|---|---|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: 2.08 mg/mL (3.57 mM) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 悬浮液;超声助溶。
例如,若需制备1 mL的工作液,可将100 μL 20.8 mg/mL澄清DMSO储备液加入400 μL PEG300中,混匀;然后向上述溶液中加入50 μL Tween-80,混匀;加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: 2.08 mg/mL (3.57 mM) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 悬浊液; 超声助溶。 例如,若需制备1 mL的工作液,可将 100 μL 20.8 mg/mL澄清DMSO储备液加入900 μL 20% SBE-β-CD生理盐水溶液中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 View More
配方 3 中的溶解度: ≥ 2.08 mg/mL (3.57 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.7154 mL | 8.5768 mL | 17.1535 mL | |
| 5 mM | 0.3431 mL | 1.7154 mL | 3.4307 mL | |
| 10 mM | 0.1715 mL | 0.8577 mL | 1.7154 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
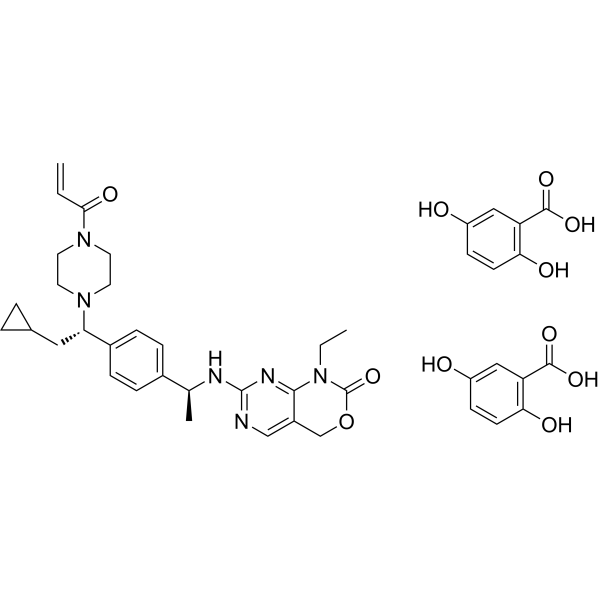 Crelosidenib (gentisate)
Crelosidenib (gentisate)
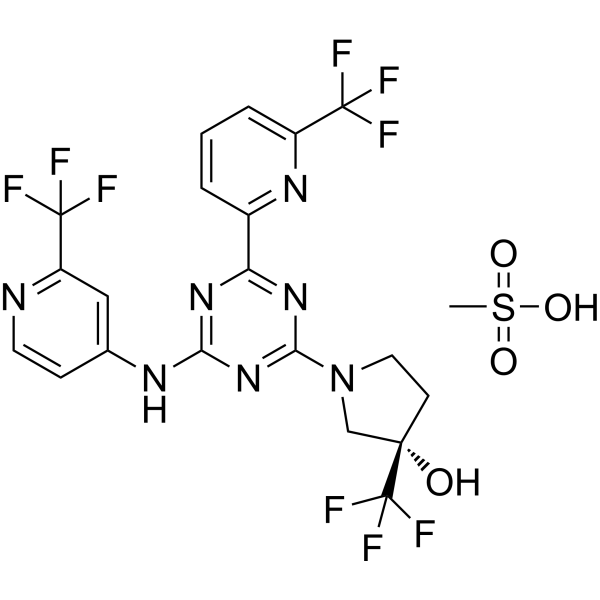 SH1573
SH1573
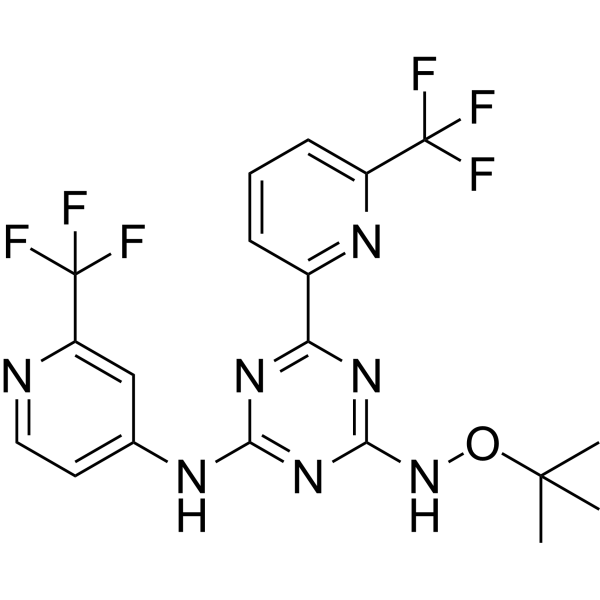 TQ05310
TQ05310
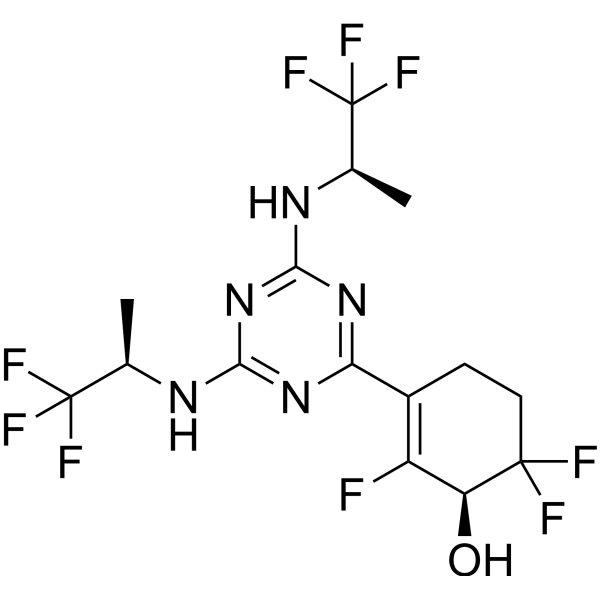 Ranosidenib
Ranosidenib
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1






























 COA
COA
