| 规格 | 价格 | |
|---|---|---|
| 500mg | ||
| 1g | ||
| Other Sizes |
| 靶点 |
β-1,3-glucan synthesis
|
|---|---|
| 体外研究 (In Vitro) |
Ibrexafungerp正在开发中,作为第一种口服和静脉注射GSI(静脉注射葡聚糖合酶抑制剂),用于治疗和预防真菌感染,包括由念珠菌属、曲霉属和支氏肺孢子虫引起的严重和危及生命的感染,有可能提供静脉注射和口服制剂的治疗优势。Ibrexafungerp导致(1→3)-β-D-葡聚糖聚合物减少,真菌细胞壁减弱。Ibrexafungerp在结构上与棘白菌素不同,与靶酶的相互作用也不同(图2)。尽管ibrexafungerp的(1→3)-β-D-葡聚糖合酶上的结合位点与棘白菌素的结合位点部分重叠,但似乎是不相同的,导致对ibrexafungarp的耐药率较低。在体外研究中,在fks突变存在的情况下,ibrexafungerp对野生型和棘白菌素耐药念珠菌菌株的活性受到的影响最小。因此,ibrexafungerp与棘白菌素的交叉抗性潜力有限。
[1]
Ibrexafungerp对一系列曲霉属分离物和念珠菌分离物具有广泛的体外活性,包括光滑念珠菌和耳念珠菌,它们表现出与棘白菌素抗真菌药耐药性相关的fks1和fks2点突变。在对氟康唑敏感性降低的念珠菌属中,包括光滑念珠菌、克鲁塞念珠菌、热带念珠菌和副丝酵母菌,使用ibrexafungerp的MIC50范围分别为0.125-1μg/mL、0.5-1μg/mL、<0.03-1μg/mL和0.25-1μ/mL。此外,正如朱[32]使用从纽约患者获得的分离物所报道的那样,ibrexafungerp(范围为0.05至0.5μg/mL)对金黄色葡萄球菌的体外活性优于氟康唑(范围为2至>256μg/mL),与棘白菌素(范围为0.015至16μg/mL)相当或更优。这一观察结果得到了其他使用全球菌株的研究的证实。Ibrexafungerp对80%的棘白菌素耐药念珠菌菌株显示出野生型MIC分布,表明fks突变对Ibrexafungerp的体外活性影响较小。[1] Ibrexafungerp(SCY-078)是一种针对葡聚糖合酶的新型一流抗真菌药物。耳念珠菌是一种新兴的多重耐药物种,已在五大洲爆发。我们采用EUCAST E.Def 7.3.1方法研究了ibrexafungerp对金黄色葡萄球菌的体外活性。白色念珠菌和光滑念珠菌,以及阿尼丁菌素、米卡芬净、两性霉素B、氟康唑、伏立康唑和伊沙唑被列为对照。对3株耳念珠菌参考菌株(CBS12372、CBS12373和CBS10913)和122株耳念珠菌、16株白色念珠菌和16株光滑念珠菌分离株进行了评估。白色念珠菌ATCC 64548、副丝酵母菌ATCC 22019和克鲁氏念珠菌ATCC 6258作为质量控制菌株。对棘球蛋白抗性分离株进行fks测序。测定MIC范围和模式MIC和MIC50值。野生型上限(野生型分布结束时的MIC上限)根据EUCAST原则确定,用于设置ECOFF。白色念珠菌和光滑念珠菌的三种QC菌株和MIC的九次重复产生了窄的MIC范围,模式MIC与已建立的EUCAST模式MIC一致,证实了稳健的测试性能。针对金黄色葡萄球菌分离物的ibrexafungerp MIC呈高斯分布,模式MIC(范围)为0.5 mg/l(0.06至2 mg/l),表明敏感性一致。在122株分离物中,8株对棘白菌素耐药,并携带S639F Fks1变异。除一例外,其余均对氟康唑耐药,伏立康唑和伊沙唑的MIC分布呈多峰分布,证实了可变敏感性。Ibrexafungerp对金黄色葡萄球菌表现出有前景的活性,包括对棘白菌素和/或其他药物具有抗性的分离株。MIC与临床和实验室标准研究所方法报告的MIC相似,表明一个共同的临床断点可能是合适的[4]。 |
| 体内研究 (In Vivo) |
SCY-078(MK-3118)是安氟霉素的一种新型半合成衍生物,是三萜类抗真菌药物中的第一种化合物。SCY-078表现出对β-(1,3)-d-葡聚糖合成的强烈抑制作用,β-(1,3,)-d-葡聚糖是许多致病真菌(包括念珠菌属和曲霉属)的重要细胞壁成分。SCY-077目前正处于治疗侵袭性真菌疾病的2期临床开发阶段。评估溶解度、肠道通透性和代谢稳定性的体外处置研究预测了良好的口服生物利用度。临床前药代动力学研究与人体每日一次给药一致。静脉注射后,啮齿动物和狗的血浆清除率较低,分别占肝血流量的<15%和<25%。啮齿动物的终末消除期半衰期为5.5至8.7小时,狗的半衰期为9.3小时。稳态下的分布体积很高(4.7至5.3升/千克),这一发现表明组织分布广泛。SCY-078在肾脏组织中的暴露量是侵袭性真菌疾病(如念珠菌病)的靶器官,在0小时至无穷大(AUC0-∞)的浓度-时间曲线下面积超过血浆20至25倍,Cmax SCY-077在多个播散性念珠菌炎小鼠模型中口服给药后达到疗效终点。药代动力学/药效学指标Cmax/MIC和AUC/MIC与结果相关。以血浆AUC0-24表示的目标治疗暴露量在各模型之间具有可比性,上限值为11.2μg·h/ml(15.4μM·h);游离药物AUC/MIC的相应平均值为∼0.75。总体而言,这些结果表明,SCY-078在播散性念珠菌病的小鼠感染模型中具有口服和静脉注射(i.v.)药代动力学特性和效力,以支持进一步研究作为侵袭性真菌疾病的新型静脉注射和口服治疗方法。[2]
耳念珠菌已被证明在住院患者中具有很高的皮肤定植风险,可能会导致医院传播。在豚鼠皮肤模型中,对动物的临床外观、组织真菌负荷、组织学和药代动力学进行了评估。与未经治疗的对照组相比,口服10mg/kg ibrexafungerp(IBX)可减轻病变的严重程度,并显著降低感染动物的耳道念珠菌真菌负担。这表明IBX有望用于控制住院患者的皮肤感染和定植[3]。 |
| 酶活实验 |
溶解性。[2]
Crystal Pharmatech测量了Ibrexafungerp/SCY-078在模拟胃液(SGF)、禁食状态模拟肠液(FaSSIF)和进食状态肠液(FeSSIF)中的溶解度。通常,将约15mg固体称重到4ml小瓶中,加入3.0ml培养基,在室温下在滚动培养箱(25rpm)上搅拌悬浮液24小时。培养后,离心0.5ml悬浮液并过滤(0.45-μm孔径),通过高压液相色谱法(HPLC)和紫外检测法测定上清液中SCY-078的浓度。根据需要调节SCY-078的量和培养基的体积,以确定其在每种培养基中的溶解度。 肝微粒体中的体外代谢稳定性。[2] 用雄性和雌性小鼠、雄性大鼠、雄性和雌性犬以及混合性别的人肝微粒体评估Ibrexafungerp/SCY-078的代谢稳定性。在NADPH存在下,将SCY-078(1μM)与混合的肝微粒体(0.5 mg蛋白质/ml)在37°C下孵育0、5、10、20和30分钟。在每个取样时间点取等分试样,用5倍体积的含有内标(d9-SCY-078125 ng/ml)的冰冷乙腈提取。根据下述条件,通过LC-MS/MS分析培养混合物中的上清液中的母体化合物。使用对照化合物7-乙氧基香豆素、普萘洛尔和维拉帕米建立微粒体制剂的代谢能力。测定了与每种物种的微粒体一起孵育的SCY-078的体外固有清除率(CLint)、CLint与体内固有清除率的比值(CL′int)和半衰期。 体外血浆蛋白结合和血液分布。[2] 通过平衡透析和液体闪烁计数法测定小鼠、大鼠、狗和人血浆中[3H]SCY-078的蛋白结合。将氚标记的Ibrexafungerp/SCY-078乙醇溶液加入到未标记的SCY-078甲醇溶液中,以制备10、100和1000μM SCY-077的恒定活性储备溶液。 在37°C下用等渗磷酸盐缓冲盐水(pH 7.4)测定蛋白质结合24小时。将每种储备溶液的等分试样(10μl)加入到混合的DBA小鼠(n=>10只动物)、Sprague-Dawley大鼠、比格犬或人血浆(1ml)中,以达到0.1、1和10μM SCY-078的最终总浓度。有机溶剂的最终浓度为0.9%(体积/体积)。在1ml丙烯酸透析细胞中进行透析,其中血浆和缓冲液由12.4-kDa截止膜分离。孵育后,对血浆和磷酸盐缓冲液的等分试样(100μl)进行放射性计数,并根据以下计算估算SCY-078的未结合分数:未结合分数=缓冲液中的放射性(dpm/0.1ml)/血浆中的总放射性(dpm/0.1ml)。 |
| 细胞实验 |
Caco2细胞单层的体外渗透性。[2]
Caco2细胞(ATCC CRL-2102)在Dulbecco改良的Eagle培养基中培养,该培养基含有二肽形式的l-谷氨酰胺(GlutaMAX)、10%(体积/体积)胎牛血清和1%(体积/容量)青霉素-链霉素,在37°C的75 ml烧瓶中,在5%CO2的加湿气氛中以10000 U/ml的浓度培养。通过在37°C下用0.25%胰蛋白酶胰蛋白酶消化5分钟来收获接近融合的Caco-2细胞培养物,并将其重新悬浮在培养基中。将细胞以约200000个细胞/cm2的密度接种到半透膜过滤器插件 上。在总共21天的培养过程中,每2至3天更换一次细胞培养基。在试验当天,用运输介质(Hanks平衡盐溶液,含25 mM葡萄糖和25 mM HEPES 冲洗细胞单层,并通过测量从顶端到基底外侧隔室的通量来评估Ibrexafungerp/SCY-078的吸收渗透。将细胞单层与SCY-078(5μM)在37°C下孵育2小时,一式三份。孵育后,从顶端和基底外侧隔室取出样品,通过LC-MS/MS测定试验化合物浓度。表观渗透系数(Papp;cm/s)计算如下:Papp=1/A·C0(dQ/dt),其中dQ/dt是基底外侧隔室内药物出现的速率(μmol/s),C0是供体隔室中的初始药物浓度(μM),A是单层的表面积(cm2)。结果表示为三份样本(n=3)的平均值±标准差。 敏感性测试。[4] EUCAST MIC是按照E.Def 7.3.1方法确定的。将Ibrexafungerp(SCY-078)纯物质在-80°C下等分储存,并在二甲亚砜(5000 mg/l)中制备储备溶液。研究的最终药物浓度范围为0.008至4mg/l。还研究了以下对照化合物(化合物来源,括号内为最终浓度范围):阿尼丁菌素(白色念珠菌和光滑念珠菌分离物为0.004至4毫克/升,金黄色念珠菌分离物的浓度为0.03至32毫克/升)、米卡芬金(白色念珠菌、光滑念珠菌分离株为0.004到4毫克/升至,金黄色梭菌分离物为0.03至32mg/升)、两性霉素B(0.004到4mg/升)、氟康唑(血液分离物为0.03-32毫克/升、金黄色念珠菌为0.5至256mg/升4毫克/升)。全程使用细胞培养处理的样品。制备2倍稀释的微量滴定板,并在使用前在-80°C下冷冻。 |
| 动物实验 |
Murine models of disseminated candidiasis. [2]
The in vivo activity of SCY-078 was evaluated using two murine models of disseminated candidiasis to establish the pharmacokinetic exposure target and PK/PD measures associated with efficacy. In the first model, the target therapeutic exposure was established across four independent experiments, each employing 7 days of twice-daily (BID) oral treatment initiated shortly after infection. To establish the PK/PD parameters associated with the outcome, the efficacy of SCY-078 was compared after single or fractionated doses starting 16 h after the infectious challenge. [2] A disseminated Candida infection was induced in C′5-deficient DBA/2N mice, weighing on average 20 g, by i.v. inoculation with C. albicans MY1055. C. albicans MY1055 was cultured on Sabouraud dextrose agar plates at 35°C for 24 h. Yeast cells were washed from the surface of agar plates into sterile saline, and the cell concentrations were quantitated by using a hemocytometer. Viable cell counts were confirmed by a serial 10-fold dilution of the cell suspension and plating on SDA plates. Plates were incubated for 24 to 48 h at 35°C, whereupon the numbers of CFU were determined. The in vitro activity of SCY-078 against the MY1055 isolate was evaluated using broth microdilution assays as described in CLSI M27-A3. The MIC endpoints were based on 50% inhibition of fungal growth at 24 h. [2] For infection, 0.2 ml of a blastospore suspension containing between 2.44 × 104 and 3.56 × 104 CFU of C. albicans MY1055 was inoculated into the lateral tail vein. Mice were housed in groups of up to 10 animals in sterile microisolator cages with sterile bedding. Water and food were provided ad lib. The infected and nonmedicated (sham-treated control) animals (n = 20) received vehicle only. Treatment groups comprised five animals each, with an additional three animals included for PK analysis for SCY-078. Blood samples were collected from infected satellite PK mice at typically 0, 0.25, 0.5, 1, 2, 4, 6, and 24 h after dose 13 on day 7. Kidneys from five mice were aseptically removed from each treatment group at day 7 after infectious challenge, unless otherwise indicated. [2] Therapy with SCY-078, caspofungin or fluconazole was initiated within 15 to 30 min after challenge. Mice were treated with SCY-078 with BID p.o. doses of 6.25, 12.5, or 25 mg/kg administered in a “fit-for-purpose” formulation. Caspofungin was administered twice daily via the intraperitoneal route at doses of 0.0078, 0.03, 0.125, and 0.5 mg/kg. Fluconazole was administered p.o. BID at doses of 0.078, 0.31, 1.25, and 5.0 mg/kg. At day 7 after challenge, the mice (n = 5/group) were euthanized, and both kidneys were aseptically removed, placed in sterile Whirl-Pak bags, weighed, and homogenized in 5 ml of sterile physiological saline. Kidney homogenates were serially 10-fold diluted in sterile saline and plated on SDA. Plates were incubated at 35°C and counted after 30 to 48 h of incubation. The CFU/g of kidney were determined, and counts from treatment groups were compared to counts from sham-treated controls using a paired two-tailed t test (Microsoft Excel). The percent clearance was determined as the number of mice with no detectable yeast, with a limit of detection of 50 yeast cells per pair of kidneys because of the dilution scheme. For data from individual mice where no detectable yeast were recovered from paired kidneys, 9.8 was entered into a Microsoft Excel spreadsheet formula [log10 (5 × raw count)/paired kidney weight)] so that the counts would be one less than the limit of detection or 49 cells per pair of kidneys. [2] Pharmacokinetic analysis was performed on samples collected from satellite-infected mice (n = 3/group) after SCY-078 dose 13 on treatment day 7. Tail bleeds were obtained (20 μl collected into 60 μl of 0.1 M sodium citrate) at time points from 15 min to 6 h, concluding with a terminal bleed at 24 h. Samples were analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS) after protein precipitation. Plasma exposure for SCY-078 was calculated from the in vitro plasma/whole-blood distribution ratio (see below). [2] Efficacy was determined in a delayed treatment model based on the protocol described above using typically 5 mice per treatment group. For the single delayed-dose treatment model, a single dose of SCY-078 was administered 16 h after infection. The target endpoint for efficacy in this model was a static effect on the tissue burden measured at 96 h posttreatment. Caspofungin and fluconazole were not evaluated in this model. In the fractionated delayed-dose treatment model, SCY-078 was administered as divided doses of either two half doses or four quarter doses relative to the single dose. The total doses were 12.5, 25, and 50 mg/kg administered as a suspension in a “fit-for-purpose” formulation. Divided doses were administered at either 0 or 48 h or at 0, 24, 48, and 72 h relative to the single dose for the half and quarter doses, respectively. Caspofungin and fluconazole were not evaluated in this model. SCY-078 pharmacokinetics in plasma and kidney tissue. [2] The pharmacokinetics and bioavailability of SCY-078 were evaluated in uninfected mouse, rat, and dog after p.o. and i.v. administration. Avastis and MPI Research are Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC)-approved facilities. Aqueous formulations of SCY-078 were prepared for p.o. or i.v. delivery, 0.45% (mass/vol) saline or 0.45% (mass/vol) saline containing 2% (mass/vol) PEG400 for i.v. administration and administered at either a 4-, 5-, or 5-ml/kg dose for mice, rats, or dogs, respectively. The formulations used for the PK studies were prepared from an optimized SCY-078 salt form that enhanced the fraction absorbed relative to the fit-for-purpose SCY-078 formulations used for the murine efficacy studies. Blood samples for PK studies were collected into tubes containing K2EDTA anticoagulant and stored on wet ice until centrifuged and processed for plasma (stored at approximately −80°C). The peak concentration (Cmax), the time to maximum concentration (Tmax), the half-life, and the AUC were determined from composite mean plasma concentration-time data for rodents and individual plasma concentration-time data for dogs. All doses and plasma concentrations of SCY-078 are presented as free base. Mice. [2] Female CD-1 mice (n = 5/sex/time point/group) weighing approximately 25 g were administered SCY-078 by oral gavage either once daily or as multiple twice-per-day (Q12h) treatments, with individual doses ranging between 3 and 100 mg/kg (6 to 150 mg/kg/day). Mice also received a single 1-mg/ml i.v. dose of SCY-078. Plasma specimens were collected prior to dosing and at 0.25, 0.5, 1, 2, 4, 6, 9, 12, 18, 24, 36, 48, and 60 h after the single i.v. dose and at the same time points from 1 h postdose onward following oral dose 13 on day 7 of treatment. On the first day of oral treatment, plasma specimens were collected immediately before each of the two doses and 1, 2, 4, 6, 9, and 12 h postdosing. Two additional samples (predose and at approximately Tmax) were collected after the first dose on day 4 of treatment to determine whether exposure had reached steady-state levels. Kidney tissues were collected at 2, 4, 12, 24, 36, 48, and 60 h after oral dose 13 on treatment day 7, blotted dry, and stored at approximately −80°C until analysis. Rats. [2] Male and female Wistar Han rats (n = 3/sex/group/time point) weighing 0.329 to 0.398 kg (males) or 0.187 to 0.244 kg (females) received a single 5-mg/kg dose of SCY-078 as an i.v. injection in the lateral tail administered over 5 min or an oral dose of 20 mg/kg as described above. Blood specimens were collected from four animals/time point/group using a sparse sampling strategy via the sublingual vein at time intervals to 72 h postdosing and processed as described above. Dogs. [2] Treatment naive male and female beagle dogs (n = 3/sex/group/time point) weighing 8 to 12 kg (males) or 6 to 10 kg (females) were given a single 5-mg/kg dose of SCY-078 as an i.v. injection over 5 min or an oral dose of 20 mg/kg as described above. Blood specimens were collected from the jugular vein at various time intervals to 72 h postdosing and processed as described above. Analysis of SCY-078 in biological samples: mouse kidney tissue homogenization. [2] On the day of analysis, either treatment-naive (drug-free) kidney tissue or freshly thawed tissue samples from PK studies were weighed and homogenized in 3 volumes of phosphate-buffered saline. The density of kidney tissue used for the determination of volume was assumed to be 1.0 g/ml. [2] Whole-kidney tissue samples were homogenized in parallel by means of a Precellys 24 bead mill homogenizer using 2-ml Precellys tubes containing 1.4-mm-diameter ceramic beads (CK14) and a single 15-s cycle of 5,500 rpm. Parallel bead homogenization increased throughput and avoided potential cross-contamination of samples since each sample was wholly contained and homogenized within its own sealed vial with no direct contact between the homogenizing instrument or other samples. |
| 药代性质 (ADME/PK) |
Absorption, Distribution and Excretion
Ibrexafungerp given at a dose of 300 mg twice daily reaches a Cmax of 435 ng/mL, with a Tmax of 4-6 hours, and an AUC0-24 of 6832 h\*ng/mL. 90% of a radiolabelled oral dose of ibrexafungerp is recovered in the feces, with 51% as the unchanged parent drug. 1% of a radiolabelled oral dose is recovered in the urine. The volume of distribution at steady state is approximately 600 L. Clearance values of 53.6 L/h and 56.1 L/h have been reported. Metabolism / Metabolites Ibrexafungerp is hydroxylated by CYP3A4 before glucuronide or sulfate conjugation of the hydroxyl group before elimination. Biological Half-Life The elimination half life of ibrexafungerp is approximately 20 hours. |
| 毒性/毒理 (Toxicokinetics/TK) |
Effects During Pregnancy and Lactation
◉ Summary of Use during Lactation No information is available on the use of ibrexafungerp during breastfeeding. The drug is over 99% protein bound, so amounts in milk are likely to be very low. If ibrexafungerp is required by the mother of an older infant, it is not a reason to discontinue breastfeeding, but until more data become available, an alternate drug may be preferred, especially while nursing a newborn or preterm infant. ◉ Effects in Breastfed Infants Relevant published information was not found as of the revision date. ◉ Effects on Lactation and Breastmilk Relevant published information was not found as of the revision date. |
| 参考文献 |
[1]. Ibrexafungerp: A Novel Oral Triterpenoid Antifungal in Development for the Treatment of Candida auris Infections. Antibiotics (Basel). 2020 Aug 25;9(9):539.
[2]. Preclinical Pharmacokinetics and Pharmacodynamic Target of SCY-078, a First-in-Class Orally Active Antifungal Glucan Synthesis Inhibitor, in Murine Models of Disseminated Candidiasis. Antimicrob Agents Chemother. 2017 Mar 24;61(4):e02068-16. [3]. Efficacy of Ibrexafungerp (SCY-078) against Candida auris in an In Vivo Guinea Pig Cutaneous Infection Model. Antimicrob Agents Chemother. 2020 Sep 21;64(10):e00854-20. [4]. In Vitro Activity of Ibrexafungerp (SCY-078) against Candida auris Isolates as Determined by EUCAST Methodology and Comparison with Activity against C. albicans and C. glabrata and with the Activities of Six Comparator Agents. Antimicrob Agents Chemother. 2020 Feb 21;64(3):e02136-19. |
| 其他信息 |
See also: Ibrexafungerp (has active moiety).
Drug Indication Treatment of invasive candidiasis |
| 分子式 |
C50H75N5O11
|
|---|---|
| 分子量 |
922.1574
|
| 精确质量 |
921.546
|
| 元素分析 |
C, 65.12; H, 8.20; N, 7.59; O, 19.08
|
| CAS号 |
1965291-08-0
|
| 相关CAS号 |
1207753-03-4;1965291-08-0 (citrate);
|
| PubChem CID |
137552087
|
| 外观&性状 |
White to off-white solid powder
|
| tPSA |
258
|
| 氢键供体(HBD)数目 |
6
|
| 氢键受体(HBA)数目 |
15
|
| 可旋转键数目(RBC) |
14
|
| 重原子数目 |
66
|
| 分子复杂度/Complexity |
1650
|
| 定义原子立体中心数目 |
12
|
| SMILES |
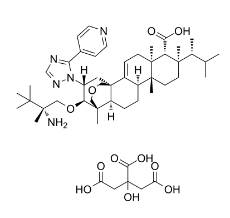
C[C@H](C(C)C)[C@]1(CC[C@@]2([C@H]3CC[C@H]4[C@]5(COC[C@]4(C3=CC[C@]2([C@@H]1C(=O)O)C)C[C@H]([C@@H]5OC[C@@](C)(C(C)(C)C)N)N6C(=NC=N6)C7=CC=NC=C7)C)C)C.C(C(=O)O)C(CC(=O)O)(C(=O)O)O
|
| InChi Key |
WKIRTJACGBEXBZ-FQGZCCSZSA-N
|
| InChi Code |
InChI=1S/C44H67N5O4.C6H8O7/c1-27(2)28(3)39(7)18-19-41(9)30-12-13-33-40(8)23-52-25-44(33,31(30)14-17-42(41,10)34(39)37(50)51)22-32(35(40)53-24-43(11,45)38(4,5)6)49-36(47-26-48-49)29-15-20-46-21-16-29;7-3(8)1-6(13,5(11)12)2-4(9)10/h14-16,20-21,26-28,30,32-35H,12-13,17-19,22-25,45H2,1-11H3,(H,50,51);13H,1-2H2,(H,7,8)(H,9,10)(H,11,12)/t28-,30+,32-,33+,34-,35+,39-,40-,41-,42+,43+,44+;/m1./s1
|
| 化学名 |
(1R,5S,6R,7R,10R,11R,14R,15S,20R,21R)-21-[(2R)-2-amino-2,3,3-trimethylbutoxy]-5,7,10,15-tetramethyl-7-[(2R)-3-methylbutan-2-yl]-20-(5-pyridin-4-yl-1,2,4-triazol-1-yl)-17-oxapentacyclo[13.3.3.01,14.02,11.05,10]henicos-2-ene-6-carboxylic acid;2-hydroxypropane-1,2,3-tricarboxylic acid
|
| 别名 |
Ibrexafungerp citrate; SCY-078 citrate; 1965291-08-0; M4NU2SDX3E; brexafemme;
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples
|
|---|---|
| 溶解度 (体内实验) |
注意: 如下所列的是一些常用的体内动物实验溶解配方,主要用于溶解难溶或不溶于水的产品(水溶度<1 mg/mL)。 建议您先取少量样品进行尝试,如该配方可行,再根据实验需求增加样品量。
注射用配方
注射用配方1: DMSO : Tween 80: Saline = 10 : 5 : 85 (如: 100 μL DMSO → 50 μL Tween 80 → 850 μL Saline)(IP/IV/IM/SC等) *生理盐水/Saline的制备:将0.9g氯化钠/NaCl溶解在100 mL ddH ₂ O中,得到澄清溶液。 注射用配方 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (如: 100 μL DMSO → 400 μL PEG300 → 50 μL Tween 80 → 450 μL Saline) 注射用配方 3: DMSO : Corn oil = 10 : 90 (如: 100 μL DMSO → 900 μL Corn oil) 示例: 以注射用配方 3 (DMSO : Corn oil = 10 : 90) 为例说明, 如果要配制 1 mL 2.5 mg/mL的工作液, 您可以取 100 μL 25 mg/mL 澄清的 DMSO 储备液,加到 900 μL Corn oil/玉米油中, 混合均匀。 View More
注射用配方 4: DMSO : 20% SBE-β-CD in Saline = 10 : 90 [如:100 μL DMSO → 900 μL (20% SBE-β-CD in Saline)] 口服配方
口服配方 1: 悬浮于0.5% CMC Na (羧甲基纤维素钠) 口服配方 2: 悬浮于0.5% Carboxymethyl cellulose (羧甲基纤维素) 示例: 以口服配方 1 (悬浮于 0.5% CMC Na)为例说明, 如果要配制 100 mL 2.5 mg/mL 的工作液, 您可以先取0.5g CMC Na并将其溶解于100mL ddH2O中,得到0.5%CMC-Na澄清溶液;然后将250 mg待测化合物加到100 mL前述 0.5%CMC Na溶液中,得到悬浮液。 View More
口服配方 3: 溶解于 PEG400 (聚乙二醇400) 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.0844 mL | 5.4221 mL | 10.8441 mL | |
| 5 mM | 0.2169 mL | 1.0844 mL | 2.1688 mL | |
| 10 mM | 0.1084 mL | 0.5422 mL | 1.0844 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
 AOH-1996
AOH-1996
 hMAO-B-IN-3
hMAO-B-IN-3
 Flupyrimin
Flupyrimin
 阿曲库铵
阿曲库铵
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1






























