
| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 10 mM * 1 mL in DMSO |
|
||
| 10mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| 500mg |
|
||
| 1g |
|
||
| Other Sizes |
|

| 靶点 |
PDE3 (IC50 = 6.5 μM); PDE4 (IC50 = 26.3 μM); PDE5 (IC50 = 31.7 μM)
|
|---|---|
| 体外研究 (In Vitro) |
KMUP-1(一种黄嘌呤肽诱导剂)和 IBMX 诱导气道松弛的最有效浓度均为 100 μM;两种化合物触发的诱导反应的激活没有明显的变化[1]。 IBMX (100 μM) 激活肾外髓质 K+ (ROMK) 通道 (n=6, P<0.05),而 ANG II (n=6, P=NS) 或 cGMP 不能进一步激活这些通道。值得注意的是,使用 IBMX (100 μM) 从高 K+ (HK) 喂养的支架中分离含有假营养物收集管 (CCD) 20 分钟后,肾小管 cAMP 含量显着增加至 1.43±0.35 pg/mm 肾小管长度 (n =14)与媒介物处理对照中测量的肾小管长度相比(0.61±0.13 pg/mm 肾小管长度,n = 12,P<0.05)[2]。
|
| 体内研究 (In Vivo) |
IBMX(一种非选择性 PDE)会显着降低肝糖原储备(mg/g,IBMX 22±1.5 P<0.001)。与血清相比,IBMX 和 mc5 被发现显着增加心脏体征(葡萄糖,mg/dl,对照=141±3,IBMX=210±17 P<0.001,mc5=191±13 P<0.01)。相反,来自 mc1、mc6、MCPIP 和 Win 47203 的化合物没有显着影响任何受试者(对照=141±3、mc1 160±7、mc6 175±9、MCPIP 179±8 和 Win 47203 116±2) P>0.05)。未发现mc2改变脐带刻度(对照=141±3和mc2=145±5)。当谈到增强心血管核心时,IBMX 是最有效的 [3]。虽然罗布麻宁和 IBMX 治疗并没有显着降低右心室 (RV) 的寒冷暴露,但它们确实大大减少了寒冷引起的收缩压升高(分别为 23.5 ± 1.8 和 24.2 ± 0.6 mmHg)。管腔直径分别增加到62.7±4.2和59.5±4.3μM,而PA内侧层的厚度分别为19.0±0.9和16.9±0.8μM][4]。
|
| 酶活实验 |
单个CCD中cAMP含量的测量。[2]
在大鼠安乐死后1小时内,在冷林格溶液中从单个肾脏中显微切割出多个单独的CCD,并将总长度约为10mm的CCD转移到1.5毫升的微量离心管中,以产生单个样品。此后,将新鲜的林格溶液(50μl)添加到每个试管中,并使样品在室温下平衡5分钟。接下来,离心样品,去除上清液,并加入含有IBMX(100μM)或载体(DMSO)的林格溶液。在室温下孵育20分钟后,对样品进行离心,并将上清液储存在−80°C下。为了测量细胞外cAMP,将上清液样品在95°C下煮沸5分钟,然后离心,将45μl上清液与5μl内标溶液混合,然后进行直接分析。为了从CCD中提取细胞内cAMP,将0.5ml冰冷的1-丙醇添加到组织颗粒中,并将样品置于4°C的冷藏室中的摇壶上2小时;1-丙醇提取物储存在−80°C下。为了分析细胞内cAMP,将1-丙醇样品干燥,在100μl纯水中重构,然后以8000rpm离心10分钟。将样品(90μl)与10μl内标溶液混合,并对其进行直接分析。cAMP测量通过HPLC串联质谱法进行,使用三重四极质谱仪,如先前详细描述的(37,38)。对于每个样品,总CCD cAMP含量计算为在单个小管的提取物中测量的cAMP含量加上在上清液中检测到的与小管长度(单位:mm)标准化的cAMP内容的总和。 |
| 细胞实验 |
细胞在24孔板中生长,每孔105个细胞。汇合时,单层细胞用磷酸盐缓冲溶液(PBS)洗涤,然后在100μM IBMX存在下与KMUP-1(0.1-100μM)孵育20分钟。通过添加10%三氯乙酸(TCA)终止孵育。对细胞悬浮液进行超声处理,然后在4°C下以2500×g离心15分钟。为了去除TCA,用5体积的水饱和乙醚提取上清液三次。然后,将上清液冻干,并通过使用市售放射免疫分析试剂盒测定每个样品的环状GMP或AMP[2]。
|
| 动物实验 |
Mice[3]
Male mice (25-35 gram) were used in the experiment. The test compounds (IBMX, MCPIP, mc1, mc2, mc5 or mc6) or solvent (control) were injected subcutaneously to mice at 1 mg/kg dosage twice a day (8:00 a.m. and 8:00 p.m.) for 7 days. Rats[4] Six groups of male Sprague-Dawley rats were used (150-180g, 6 rats/group). Three groups of rats are exposed to a climate-controlled walk-in chamber maintained at moderate cold (5.0±1°C). The remaining groups are kept in an identical chamber maintained at room temperature (23.5±1°C, warm) and served as controls. After eight weeks of exposure to cold, 3 groups in each temperature condition received continuous IV infusion of IBMX (PDE-1 inhibitor, 8.5 mg/kg/day), Apocynin (NADPH oxidase inhibitor, 25 mg/kg/day) and vehicle (DMSO, 50%), respectively. The doses of drugs have been validated for effective inhibition of PDE-1 and NADPH oxidase activity, respectively. Body weight is measured weekly. After one week of drug infusion, the animals’ right ventricular systolic blood pressure (RVBP) is measured under anesthesia. The RVP is a reliable indicator of pulmonary arterial blood pressure (PAP) and has been used by numerous investigators for evaluating PH. |
| 毒性/毒理 (Toxicokinetics/TK) |
mouse LD50 intraperitoneal 44 mg/kg European Journal of Medicinal Chemistry--Chimie Therapeutique., 25(653), 1990
|
| 参考文献 |
|
| 其他信息 |
3-isobutyl-1-methyl-9H-xanthine is a 3-isobutyl-1-methylxanthine. It is functionally related to a 9H-xanthine. It is a tautomer of a 3-isobutyl-1-methyl-7H-xanthine.
A potent cyclic nucleotide phosphodiesterase inhibitor; due to this action, the compound increases cyclic AMP and cyclic GMP in tissue and thereby activates CYCLIC NUCLEOTIDE-REGULATED PROTEIN KINASES. 7-[2-[4-(2-chlorophenyl)piperazinyl]ethyl]-1,3-dimethylxanthine (KMUP-1) produces tracheal relaxation, intracellular accumulation of cyclic nucleotides, inhibition of phosphodiesterases (PDEs) and activation of K+ channels. KMUP-1 (0.01-100 microm) induced concentration-dependent relaxation responses in guinea-pig epithelium-intact trachea precontracted with carbachol. Relaxation responses were also elicited by the PDE inhibitors theophylline, 3-isobutyl-1-methylxanthine (IBMX), milrinone, rolipram and zaprinast (100 microm), and a KATP channel opener, levcromakalim. Tracheal relaxation induced by KMUP-1 was attenuated by epithelium removal and by pretreatment with inhibitors of soluble guanylate cyclase (sGC) (1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ), 1 microm), nitric oxide synthase (Nomega-nitro-L-arginine methyl ester, 100 microm), K+ channels (tetraethylammonium, 10 mm), KATP channels (glibenclamide, 1 microm), voltage-dependent K+ channels (4-aminopyridine, 100 microm) and Ca2+-dependent K+ channels (charybdotoxin, 0.1 microm or apamin, 1 microm). Both KMUP-1 (10 microm) and theophylline nonselectively and slightly inhibited the enzyme activity of PDE3, 4 and 5, suggesting that they are able to inhibit the metabolism of adenosine 3',5'-cyclic monophosphate (cyclic AMP) and guanosine 3',5'-cyclic monophosphate (cyclic GMP). Likewise, the effects of IBMX were also measured and its IC50 values for PDE3, 4 and 5 were 6.5 +/- 1.2, 26.3 +/- 3.9 and 31.7 +/- 5.3 microm, respectively. KMUP-1 (0.01-10 microm) augmented intracellular cyclic AMP and cyclic GMP levels in guinea-pig cultured tracheal smooth muscle cells. These increases in cyclic AMP and cyclic GMP were abolished in the presence of an adenylate cyclase inhibitor SQ 22536 (100 microm) and an sGC inhibitor ODQ (10 microm), respectively. KMUP-1 (10 microm) increased the expression of protein kinase A (PKARI) and protein kinase G (PKG1alpha1beta) in a time-dependent manner, but this was only significant for PKG after 9 h. Intratracheal administration of tumour necrosis factor-alpha (TNF-alpha, 0.01 mg kg(-1)) induced bronchoconstriction and exhibited a time-dependent increase in lung resistance (RL) and decrease in dynamic lung compliance (Cdyn). KMUP-1 (1.0 mg kg(-1)), injected intravenously for 10 min before the intratracheal TNF-alpha, reversed these changes in RL and Cdyn. These data indicate that KMUP-1 activates sGC, produces relaxation that was partly dependent on an intact epithelium, inhibits PDEs and increases intracellular cyclic AMP and cyclic GMP, which then increases PKA and PKG, leading to the opening of K+ channels and resulting tracheal relaxation.[1] The kidney adjusts K⁺ excretion to match intake in part by regulation of the activity of apical K⁺ secretory channels, including renal outer medullary K⁺ (ROMK)-like K⁺ channels, in the cortical collecting duct (CCD). ANG II inhibits ROMK channels via the ANG II type 1 receptor (AT1R) during dietary K⁺ restriction. Because AT1Rs and ANG II type 2 receptors (AT2Rs) generally function in an antagonistic manner, we sought to characterize the regulation of ROMK channels by the AT2R. Patch-clamp experiments revealed that ANG II increased ROMK channel activity in CCDs isolated from high-K⁺ (HK)-fed but not normal K⁺ (NK)-fed rats. This response was blocked by PD-123319, an AT2R antagonist, but not by losartan, an AT1R antagonist, and was mimicked by the AT2R agonist CGP-42112. Nitric oxide (NO) synthase is present in CCD cells that express ROMK channels. Blockade of NO synthase with N-nitro-l-arginine methyl ester and free NO with 2-(4-carboxyphenyl)-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide potassium salt completely abolished ANG II-stimulated ROMK channel activity. NO enhances the synthesis of cGMP, which inhibits phosphodiesterases (PDEs) that normally degrade cAMP; cAMP increases ROMK channel activity. Pretreatment of CCDs with IBMX, a broad-spectrum PDE inhibitor, or cilostamide, a PDE3 inhibitor, abolished the stimulatory effect of ANG II on ROMK channels. Furthermore, PKA inhibitor peptide, but not an activator of the exchange protein directly activated by cAMP (Epac), also prevented the stimulatory effect of ANG II. We conclude that ANG II acts at the AT2R to stimulate ROMK channel activity in CCDs from HK-fed rats, a response opposite to that mediated by the AT1R in dietary K⁺-restricted animals, via a NO/cGMP pathway linked to a cAMP-PKA pathway.[2] Objective(s): PDE3 has a functional role in insulin secretion and action. We investigated the metabolic effects of new synthetic PDE3 inhibitors (mc1, mc2, mc5 and mc6), on mice and hyperglycemic rat. Materials and methods: The test compound or solvent was injected subcutaneously to mice, for 7 days. On day 8, blood and liver samples were obtained. In hyperglycemic rat, 0.5 g/kg glucose with or without test compounds was injected, and followed with infusion of 1.5 g/kg/hr glucose. Blood samples were collected in mentioned intervals and liver was dissected. Results: In hyperglycemic rat, all test compounds decreased blood glucose and the effect of milrinone was potentiated by glybenclamide. Milrinone or IBMX did not change plasma insulin levels, but it was augmented by combination of milrinone and glybenclamide. In both species, liver glycogen storage was decreased by IBMX, mc5, mc6 or MCPIP, increased by mc2 (liver glycogen, rat, control=56±2, mc2=70±3 P< 0.01, mice, control=33±0.7, mc2=42±2.3 P< 0.01) and was not changed in the presence of mc1. Milrinone did not change the glycogen storage in rats though increased it in mice (control= 33±0.7, milrinone= 40±1 P< 0.05). Conclusion: Increasing plasma insulin levels by combination of milrinone and glybenclamide confirmed that in hyperglycemic rat, the hypoglycemic effect was correlated with increasing insulin secretion. Variations of plasma insulin were obscured by the pulsative characteristic of pancreatic insulin release. Decreasing glycogen storage reflected inhibition of liver PDE activity. The reasons for ineffectiveness of mc1, anabolic effect of mc2, and differential effects of milrinone were not clear. [3] Chronic exposure to cold caused pulmonary arterial hypertension (cold-induced pulmonary hypertension [CIPH]) and increased phosphodiesterase-1C (PDE-1C) expression in pulmonary arteries (PAs) in rats. The purpose of this study is to investigate a hypothesis that inhibition of PDE-1 would decrease inflammatory infiltrates and superoxide production leading to attenuation of CIPH. Three groups of male rats were exposed to moderate cold (5±1°C) continuously, whereas 3 groups were maintained at room temperature (23.5±1°C, warm; 6 rats/group). After 8-week exposure to cold, 3 groups in each temperature condition received continuous intravenous infusion of 8-isobutyl-methylxanthine (8-IBMX) (PDE-1 inhibitor), apocynin (NADPH oxidase inhibitor) or vehicle, respectively, for 1 week. Cold exposure significantly increased right-ventricular systolic pressure compared with warm groups (33.8±3.2 versus 18.6±0.3 mm Hg), indicating that animals developed CIPH. Notably, treatment with 8-IBMX significantly attenuated the cold-induced increase in right ventricular pressure (23.5±1.8 mm Hg). Cold exposure also caused right-ventricular hypertrophy, whereas 8-IBMX reversed cold-induced right ventricular hypertrophy. Cold exposure increased PDE-1C protein expression, macrophage infiltration, NADPH oxidase activity, and superoxide production in PAs and resulted in PA remodeling. 8-IBMX abolished cold-induced upregulation of PDE-1C in PAs. Interestingly, inhibition of PDE-1 eliminated cold-induced macrophage infiltration, NADPH oxidase activation, and superoxide production in PAs and reversed PA remodeling. Inhibition of NADPH oxidase by apocynin abolished cold-induced superoxide production and attenuated CIPH and PA remodeling. In conclusion, inhibition of PDE-1 attenuated CIPH and reversed cold-induced PA remodeling by suppressing macrophage infiltration and superoxide production, suggesting that upregulation of PDE-1C expression may be involved in the pathogenesis of CIPH. [4] |
| 分子式 |
C10H14N4O2
|
|
|---|---|---|
| 分子量 |
222.25
|
|
| 精确质量 |
222.111
|
|
| 元素分析 |
C, 54.04; H, 6.35; N, 25.21; O, 14.40
|
|
| CAS号 |
28822-58-4
|
|
| 相关CAS号 |
IBMX;28822-58-4
|
|
| PubChem CID |
3758
|
|
| 外观&性状 |
White to light yellow sosild
|
|
| 密度 |
1.3±0.1 g/cm3
|
|
| 沸点 |
445.6±37.0 °C at 760 mmHg
|
|
| 熔点 |
200-201 °C(lit.)
|
|
| 闪点 |
223.3±26.5 °C
|
|
| 蒸汽压 |
0.0±1.1 mmHg at 25°C
|
|
| 折射率 |
1.569
|
|
| LogP |
1.24
|
|
| tPSA |
72.68
|
|
| 氢键供体(HBD)数目 |
1
|
|
| 氢键受体(HBA)数目 |
3
|
|
| 可旋转键数目(RBC) |
2
|
|
| 重原子数目 |
16
|
|
| 分子复杂度/Complexity |
318
|
|
| 定义原子立体中心数目 |
0
|
|
| SMILES |
O=C1N(C([H])([H])[H])C(C2=C(N=C([H])N2[H])N1C([H])([H])C([H])(C([H])([H])[H])C([H])([H])[H])=O
|
|
| InChi Key |
APIXJSLKIYYUKG-UHFFFAOYSA-N
|
|
| InChi Code |
InChI=1S/C10H14N4O2/c1-6(2)4-14-8-7(11-5-12-8)9(15)13(3)10(14)16/h5-6H,4H2,1-3H3,(H,11,12)
|
|
| 化学名 |
|
|
| 别名 |
|
|
| HS Tariff Code |
2934.99.9001
|
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
|
|||
|---|---|---|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 1.67 mg/mL (7.51 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 16.7 mg/mL澄清的DMSO储备液加入到400 μL PEG300中,混匀;再向上述溶液中加入50 μL Tween-80,混匀;然后加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: 1.67 mg/mL (7.51 mM) (饱和度未知) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 悬浊液; 超声助溶。 例如,若需制备1 mL的工作液,可将 100 μL 16.7mg/mL澄清的DMSO储备液加入到900μL 20%SBE-β-CD生理盐水中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 View More
配方 3 中的溶解度: ≥ 1.67 mg/mL (7.51 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 配方 4 中的溶解度: ≥ 0.71 mg/mL (3.19 mM) (饱和度未知) in 10% EtOH + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将100 μL 7.1 mg/mL 澄清 EtOH 储备液加入400 μL PEG300 中,混匀;再向上述溶液中加入50 μL Tween-80,混匀;然后加入450 μL 生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 5 中的溶解度: ≥ 0.71 mg/mL (3.19 mM) (饱和度未知) in 10% EtOH + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,将 100 μL 7.1 mg/mL 澄清乙醇储备液加入到 900 μL 20% SBE-β-CD 生理盐水溶液中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 配方 6 中的溶解度: ≥ 0.71 mg/mL (3.19 mM) (饱和度未知) in 10% EtOH + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 7.1 mg/mL 澄清 EtOH 储备液添加到 900 μL 玉米油中并充分混合。 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 4.4994 mL | 22.4972 mL | 44.9944 mL | |
| 5 mM | 0.8999 mL | 4.4994 mL | 8.9989 mL | |
| 10 mM | 0.4499 mL | 2.2497 mL | 4.4994 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
|
Total cAMP content in single CCDs of HK-fed rats is increased after IBMX treatment.Am J Physiol Renal Physiol.2014 Oct 1;307(7):F833-43 |
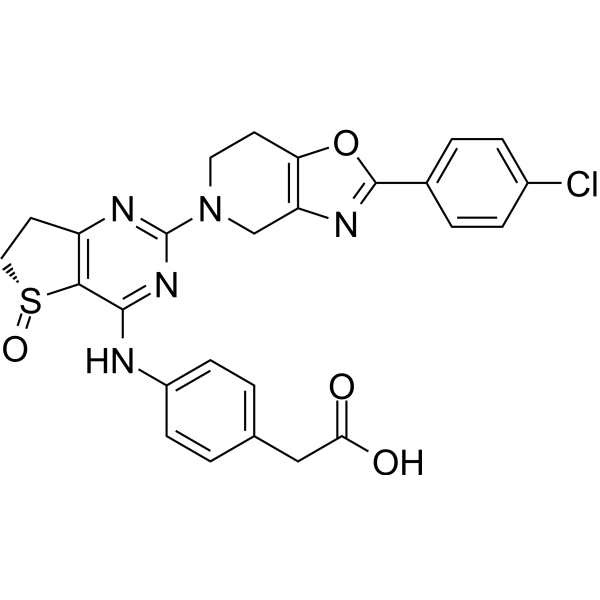 PDE4B/D-IN-1
PDE4B/D-IN-1
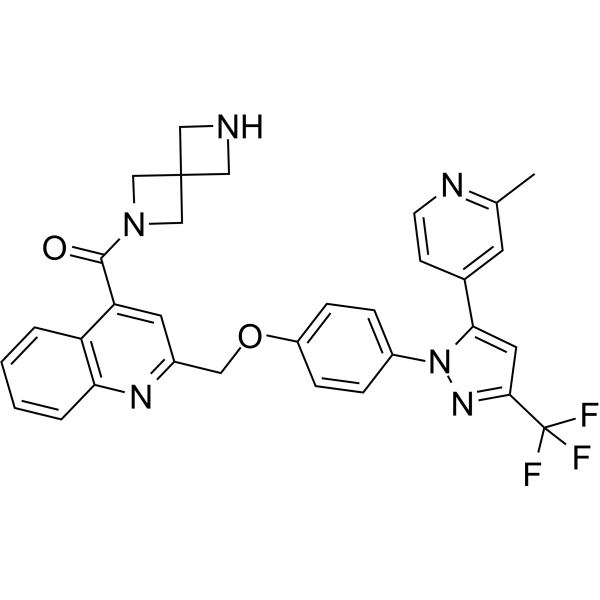 Phosphodiesterase-IN-2
Phosphodiesterase-IN-2
 PDE4B/D-IN-3
PDE4B/D-IN-3
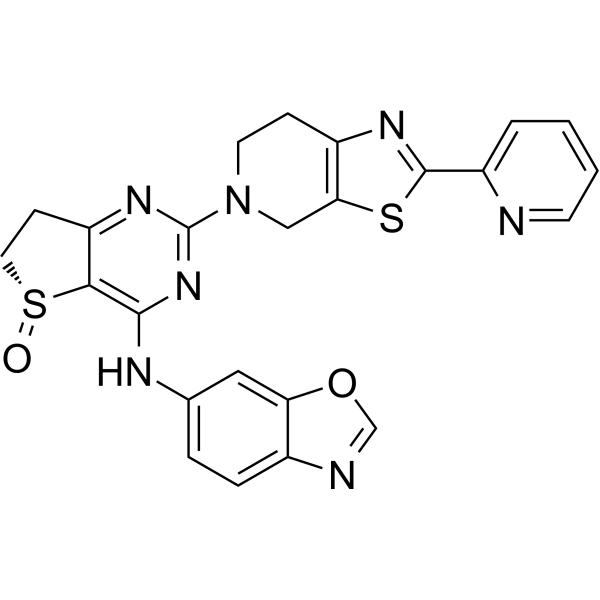 PDE4B/D-IN-2
PDE4B/D-IN-2
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1





























 COA
COA


