
| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 1g |
|
||
| 2g |
|
||
| 5g |
|
||
| 10g |
|
||
| 25g |
|
||
| Other Sizes |
|

| 靶点 |
D2 Receptor
|
|
|---|---|---|
| 体外研究 (In Vitro) |
氟哌啶醇是一种由中心哌啶结构和N-连接的对氟丁苯部分组成的化合物,中心哌啶结构在4位具有羟基和对氯苯基取代基。它具有5-羟色胺能拮抗剂、第一代抗精神病药、多巴胺能拮抗剂和止吐药的作用。它是一种羟基哌啶、有机氟化合物、芳香酮、叔醇和一氯苯类化合物。
|
|
| 体内研究 (In Vivo) |
1.在离体血液灌注犬胰腺的制备中,研究了DOPA脱羧酶抑制剂、多巴胺β羟化酶和单胺氧化酶抑制剂以及氟哌啶醇对L-DOPA和多巴胺诱导的胰液分泌的影响。DOPA脱羧酶抑制剂Ro 4-4602(300 mug)完全拮抗了输注L-DOPA(100 mug/min)引起的分泌增加。动脉内多巴胺(1-10mug)的促分泌作用不受Ro 4-4602的影响,但通过输注多巴胺β羟化酶抑制剂富马酸(100mug/min)而增强。静脉注射单胺氧化酶抑制剂烟酰胺(100mg/kg)可增强多巴胺(1-10mug)诱导的分泌增加。动脉内注射氟哌啶醇(1mg)可减弱多巴胺诱导的胰腺分泌。结论是L-DOPA在腺泡细胞中转化为多巴胺,导致胰液分泌增加,因此多巴胺的细胞内水平可能受到酶平衡的控制。[2]
动脉注射氟哌啶醇 (1 mg) 可减弱多巴胺诱导的胰腺分泌。氟哌啶醇 (3 mg) 完全抑制狗胰腺中 10 μg 多巴胺的作用[1]。给小鼠注射 50 mg/kg (2 µc) 麦司卡林 45 分钟后,氟哌啶醇 (10 mg/kg) 以及氯丙嗪 (CPZ,15 mg/kg) 可在 7 至 10 分钟内阻断麦司卡林诱导的行为改变。氟哌啶醇对麦司卡林消失没有影响[2]。 |
|
| 动物实验 |
|
|
| 药代性质 (ADME/PK) |
Absorption, Distribution and Excretion
Haloperidol is a highly lipophilic compound and is extensively metabolized in humans, which may cause a large interindividual variability in its pharmacokinetics. Studies have found a wide variance in pharmacokinetic values for orally administered haloperidol with 1.7-6.1 hours reported for time to peak plasma concentration (tmax), 14.5-36.7 hours reported for half-life (t1⁄2), and 43.73 μg/L•h [range 14.89-120.96 μg/L•h] reported for AUC. Haloperidol is well-absorbed from the gastrointestinal tract when ingested orally, however, the first-pass hepatic metabolism decreases its oral bioavailability to 40 - 75%. After intramuscular administration, the time to peak plasma concentration (tmax) is 20 minutes in healthy individuals or 33.8 minutes in patients with schizophrenia, with a mean half-life of 20.7 hours. Bioavailability following intramuscular administration is higher than that for oral administration. Administration of haloperidol decanoate (the depot form of haloperidol for long-term treatment) in sesame oil results in slow release of the drug for long-term effects. The plasma concentrations of haloperidol gradually rise, reaching its peak concentration at about 6 days after the injection, with an apparent half-life of about 21 days. Steady-state plasma concentrations are achieved after the third or fourth dose. In radiolabeling studies, approximately 30% of the radioactivity is excreted in the urine following a single oral administration of 14C-labelled haloperidol, while 18% is excreted in the urine as haloperidol glucuronide, demonstrating that haloperidol glucuronide is a major metabolite in the urine as well as in plasma in humans. The apparent volume of distribution was found to range from 9.5-21.7 L/kg. This high volume of distribution is in accordance with its lipophilicity, which also suggests free movement through various tissues including the blood-brain barrier. Following intravenous administration, the plasma or serum clearance (CL) was found to be 0.39-0.708 L/h/kg (6.5 to 11.8 ml/min/kg). Following oral administration, clearance was found to be 141.65 L/h (range 41.34 to 335.80 L/h). Haloperidol clearance after extravascular administration ranges from 0.9-1.5 l/h/kg, however this rate is reduced in poor metabolizers of C_YP2D6_ enzyme. Reduced CYP2D6 enzyme activity may result in increased concentrations of haloperidol. The inter-subject variability (coefficient of variation, %) in haloperidol clearance was estimated to be 44% in a population pharmacokinetic analysis in patients with schizophrenia. Genetic polymorphism of CYP2D6 has been demonstrated to be an important source of inter-patient variability in the pharmacokinetics of haloperidol and may affect therapeutic response and incidence of adverse effects. Haloperidol is well absorbed from the gastrointestinal tract but first-pass hepatic metabolism decreases oral bioavailability to 40 to 75%. Serum concentration peaks 0.5 to 4 hours after an oral dose. The apparent volume of distribution is about 20 L/kg, consistent with the high lipophilicity of the drug. Haloperidol circulates in blood bound predominantly (90-94%) to plasma proteins. Following administration of haloperidol in animals, the drug is distributed mainly into the liver, with lower concentrations being distributed into the brain, lung, kidneys, spleen, and heart. ... Haloperidol is about 92% bound to plasma proteins. Metabolism / Metabolites Haloperidol is extensively metabolised in the liver with only about 1% of the administered dose excreted unchanged in urine. In humans, haloperidol is biotransformed to various metabolites, including p-fluorobenzoylpropionic acid, 4-(4-chlorophenyl)-4-hydroxypiperidine, reduced haloperidol, pyridinium metabolites, and haloperidol glucuronide. In psychiatric patients treated regularly with haloperidol, the concentration of haloperidol glucuronide in plasma is the highest among the metabolites, followed, in rank order, by unchanged haloperidol, reduced haloperidol and reduced haloperidol glucuronide. The drug is thought to be metabolized primarily by oxidative N-dealkylation of the piperidine nitrogen to form fluorophenylcarbonic acids and piperidine metabolites (which appear to be inactive), and by reduction of the butyrophenone carbonyl to the carbinol, forming _hydroxyhaloperidol_. The enzymes involved in the biotransformation of haloperidol include cytochrome P450 (CYP) including CYP3A4 and CYP2D6, carbonyl reductase and uridine di-phosphoglucose glucuronosyltransferase enzymes. The greatest proportion of the intrinsic hepatic clearance of haloperidol is performed by glucuronidation and followed by the reduction of haloperidol to reduced haloperidol and by CYP-mediated oxidation. In studies of cytochrome-mediated disposition in vitro, CYP3A4 appears to be the major isoform of the enzyme responsible for the metabolism of haloperidol in humans. The intrinsic clearance of the back-oxidation of reduced haloperidol to the parent compound, oxidative N-dealkylation and pyridinium formation are of the same order of magnitude. This suggests that the same enzyme system is responsible for the above three metabolic reactions. In vivo human studies on haloperidol metabolism have shown that the glucuronidation of haloperidol accounts for 50 to 60% of haloperidol biotransformation and that approximately 23% of the biotransformation was accounted for by the reduction pathway. The remaining 20 to 30% ofthe biotransformation of haloperidol would be via N-dealkylation and pyridinium formation. Although the exact metabolic fate has not been clearly established, it appears that haloperidol is principally metabolized in the liver. The drug appears to be metabolized principally by oxidative N-dealkylation of the piperidine nitrogen to form fluorophenylcarbonic acids and piperidine metabolites (which appear to be inactive), and by reduction of the butyrophenone carbonyl to the carbinol, forming hydroxyhaloperidol. Limited data suggest that the reduced metabolite, hydroxyhaloperidol, has some pharmacologic activity, although its activity appears to be less than that of haloperidol. Urinary metabolites in rats include p-fluorophenaceturic acid, beta-p-fluorobenzoylpropionic acid, and several unidentified acids. ... it is metabolized via reduction to reduced haloperidol, which is biologically inactive. Different extents of enterohepatic recycling, and ethnic differences in metabolism, could also account for the observed variability in haloperidol disposition. The enzymes involved in the biotransformation of haloperidol include cytochrome P450 (CYP), carbonyl reductase and uridine diphosphoglucose glucuronosyltransferase. The greatest proportion of the intrinsic hepatic clearance of haloperidol is by glucuronidation, followed by the reduction of haloperidol to reduced haloperidol and by CYP-mediated oxidation. In studies of CYP-mediated disposition in vitro, CYP3A4 appears to be the major isoform responsible for the metabolism of haloperidol in humans. The intrinsic clearances of the back-oxidation of reduced haloperidol to the parent compound, oxidative N-dealkylation and pyridinium formation are of the same order of magnitude, suggesting that the same enzyme system is responsible for the 3 reactions. Large variation in the catalytic activity was observed in the CYP-mediated reactions, whereas there appeared to be only small variations in the glucuronidation and carbonyl reduction pathways. Haloperidol is a substrate of CYP3A4 and an inhibitor, as well as a stimulator, of CYP2D6. ... In vivo pharmacogenetic studies have indicated that the metabolism and disposition of haloperidol may be regulated by genetically determined polymorphic CYP2D6 activity. However, these findings appear to contradict those from studies in vitro with human liver microsomes and from studies of drug interactions in vivo. Interethnic and pharmacogenetic differences in haloperidol metabolism may explain these observations. Haloperidol has known human metabolites that include p-Fluorobenzoylpropionic acid and 4-(4-chlorophenyl)-4-hydroxypiperidine, (2S,3S,4S,5R)-6-[4-(4-Chlorophenyl)-1-[4-(4-fluorophenyl)-4-oxobutyl]piperidin-4-yl]oxy-3,4,5-trihydroxyoxane-2-carboxylic acid, and Haloperidol pyridinium. Haloperidol is a known human metabolite of reduced_haloperidol. Haloperidol is well absorbed from the gastrointestinal tract but first-pass hepatic metabolism decreases oral bioavailability to 40 to 75%. Serum concentration peaks 0.5 to 4 hours after an oral dose. Following administration of haloperidol, the drug is distributed mainly into the liver, with lower concentrations being distributed into the brain, lung, kidneys, spleen, and heart. Although the exact metabolic fate has not been clearly established, it appears that haloperidol is principally metabolized in the liver. The drug appears to be metabolized principally by oxidative N-dealkylation of the piperidine nitrogen to form fluorophenylcarbonic acids and piperidine metabolites (which appear to be inactive), and by reduction of the butyrophenone carbonyl to the carbinol, forming hydroxyhaloperidol. Limited data suggest that the reduced metabolite, hydroxyhaloperidol, has some pharmacologic activity, although its activity appears to be less than that of haloperidol. Urinary metabolites include p-fluorophenaceturic acid, beta-p-fluorobenzoylpropionic acid, and several unidentified acids (A637, A566, A637). Half Life: 3 weeks Biological Half-Life Following oral administration, the half-life was found to be 14.5-36.7 hours. Following intramuscular injection, mean half-life was found to be 20.7 hours. 10 MG Haloperidol IV and oral administration to healthy volunteers: serum T1/2 10-19 hr after IV and 12-38.3 hr after oral administration. Bioavailability in the order of 60%; distribution volume around 1300 L. Haloperidol, Elimination: Oral: 24 hours (range 12 to 37 hours). Intramuscular: 21 hours (range, 17 to 25 hours). Intravenous: 14 hours (range, 10 to 19 hours). Haloperidol decanoate, Elimination: Approximately 3 weeks (single or multiple doses). |
|
| 毒性/毒理 (Toxicokinetics/TK) |
Toxicity Summary
IDENTIFICATION: Haloperidol is an antipsychotic agent. Haloperidol is a synthetic product. Haloperidol is the first of the butyrophenone series of major tranquillizers. Haloperidol is indicated for use in the management of manifestations of psychotic disorders such as schizophrenia and mania. It is indicated for the control of tics and vocal utterances of Tourette's Disorder in children and adults. It is effective for the treatment of severe behaviour problems in children of combative, explosive hyperexcitability. It is also used in the management of Gilles de La Tourette's syndrome, intractable hiccup and as an anti-emetic. HUMAN EXPOSURE: Main risks and target organs: The main features of severe overdosage are extrapyramidal reactions, hypotension, respiratory difficulty and impairment of consciousness. Haloperidol acts mainly as a dopamine antagonist. Summary of clinical effects: Consciousness may be depressed, progressing to coma; paradoxically, some patients manifest confusion, excitement and restlessness. Tremor or muscle twitching, muscle spasm, rigidity and convulsions are seen. Extrapyramidal signs can include dystonia, sometimes severe enough to impair swallowing or breathing; torticollis, oculogyric crises and opisthotonos. The pupils may be constricted or dilated. Hypotension and tachycardia are common. Sometimes there can be cardiac arrhythmias, including ventricular fibrillation, conduction defects and cardiac arrest. Contraindications: Severe dystonic reactions have followed the use of haloperidol, particularly in children and adolescents. It should therefore be used with extreme care in children. Haloperidol may also cause severe neurotoxic reactions in patients with hyperthyroidism and in patients receiving lithium. Haloperidol is contraindicated in severe toxic central nervous system depression or comatose states from any cause and individuals who are hypersensitive to this drug or have Parkinson's disease. Also contraindicated in late pregnancy because of dystonic reaction in the neonate. Infants should not be nursed during drug treatment. Routes of entry: Oral: It is the main route of administration. Parenteral: Through intravenous and intramuscular injection. Absorption by route of exposure: Haloperidol is readily absorbed from the gastrointestinal tract. Owing to the first-pass effect of metabolism in the liver, plasma concentrations following oral administration are lower than those following intramuscular administration. There is wide intersubject variation in plasma concentration of haloperidol and its therapeutic effects. The decanoate ester of haloperidol is very slowly absorbed from the site of injection and is therefore suitable for depot injection. It is gradually released into the bloodstream where it is rapidly hydrolysed to haloperidol. Distribution by route of exposure: Haloperidol is very extensively bound to plasma proteins (90%). It is widely distributed in body and crosses the bloodbrain barrier. Biological half-life by route of exposure: The plasma half-life in therapeutic doses is reported to range from about 13 to nearly 40 hours (Reynolds, 1989), with a mean of 20 hours. Metabolism: Haloperidol is metabolized in the liver and the paths of metabolism include oxidative N-dealkylation. Elimination by route of exposure: This rate of total systemic clearance increases in children and decreases in aged patients. After metabolism, haloperidol is excreted in the urine, via the bile and in the feces, there is evidence of enterohepatic recycling by 40%. About 26% was excreted in the urine by the healthy subjects and 20% by the patients in the first 5 days; by the third day about 15% had been excreted in the feces. It takes 28 days to fully eliminate a single oral dose. Mode of action: Pharmacodynamics: Dopamine receptors currently are classified as D-1(stimulate adenylate cyclase) and D-2(inhibit adenylate cyclase). Neuroleptic drugs block both D-1 and D-2 receptors but the significance of the ratio remains unclear. The therapeutic dose of neuroleptic drug appears to correlate with its affinity for brain dopamine D-2 receptors. Neuroleptic drugs also block a number of other receptors including H1 and H2 histamine, alfa 1 and alfa 2 adrenergic, muscarinic and serotoninergic receptors. Toxicity: Human data: Three cases of sudden death after taking 20 to 140 mg daily for one to four days. Children: A 29-month-old girl and an 11 month old boy who divided 265 mg of haloperidol between them developed lethargy, hypothermia, hyperreflexia, neuromuscular rigidity, unsteady gait and intention tremors. Although disturbances such as galactorrhea, amenorrhea, gynaecomastia, and impotence have been reported, the clinical significance of elevated serum prolactin levels is unknown for most patients. There are no well controlled studies with haloperidol in pregnant women. There are reports, however, of cases of limb malformations observed following maternal use of haloperidol along with other drugs which have suspected teratogenic potential during the first trimester of pregnancy. Causal relationships were not established in these cases. Since such experience does not exclude the possibility of fetal damage due to haloperidol; this drug should be used during pregnancy or in women likely to become pregnant only if the benefit clearly justifies a potential risk to the fetus. Interactions: The use of alcohol with this drug should be avoided due to possible additive effects and hypotension. An encephalopathic syndrome (characterised by weakness, lethargy, fever, tremulousness and confusion, extrapyramidal symptoms, leucocytosis, elevated serum enzymes, BUN, and FBS) followed by irreversible brain damage has occurred in a few patients treated with lithium plus haloperidol. A casual relationship between these events and the concomitant administration of lithium and haloperidol has not been established; however, patients receiving such combination therapy should be monitored closely for early evidence of neurological toxicity and treatment discontinued promptly if such signs appear (Physician's Desk Reference, 1987). Other reported interactions involve the following drugs and adverse effects: Beta-blockers: Severe hypotension or pulmonary arrest. Methyldopa: Dementia, psychomotor retardation, memory impairment and inability to concentrate. Indomethacin: Severe drowsiness and confusion. Main adverse effects: In general, the symptoms of overdose would be an exaggeration of known pharmacological effects and adverse reactions. Anticholinergic side effects and sedation occur less often than with aliphatic phenothiazines, but extrapyramidal reactions are more common. Administration of antidopaminergic and anticholinergics may worsen or bring forward the onset of extrapyramidal effects. Idiosyncratic reaction producing severe drowsiness when used with indomethacin. ANIMAL/PLANT STUDIES: Carcinogenicity: Carcinogenicity studies using oral haloperidol were conducted in Wistar rats and in Albino Swiss mice. In the rat study, survival was less than optimal in all dose groups, reducing the number of rats at risk for developing tumors. However, although a relatively greater number of rats survived to the end of the study in high dose male and female groups, these animals did not have a greater incidence of tumors than control animals. Therefore, although not optimal, this study does suggest the absence of haloperidol related increase in the incidence of neoplasia in rats. In female mice there was a statistically significant increase in mammary gland neoplasia and total tumor incidence; there was a statistically significant increase in pituitary gland neoplasia. In male mice, no statistically significant differences in incidence of total tumors or specific tumor types were noted. Neuroleptic drugs elevate prolactin levels; the elevation persists during chronic administration. Teratogenicity: Rodents haloperidol by oral or parenteral routes showed an increase in incidence of resorption, reduced fertility, delayed delivery and pup mortality. No teratogenic effect has been reported in rats, rabbits or dogs at dosages within this range, but cleft palate has been observed in mice. Mutagenicity: No mutagenic potential of haloperidol was found in the Ames Salmonella microsomal activation assay. The precise mechanism whereby the therapeutic effects of haloperidol are produced is not known, but the drug appears to depress the CNS at the subcortical level of the brain, midbrain, and brain stem reticular formation. Haloperidol seems to inhibit the ascending reticular activating system of the brain stem (possibly through the caudate nucleus), thereby interrupting the impulse between the diencephalon and the cortex. The drug may antagonize the actions of glutamic acid within the extrapyramidal system, and inhibitions of catecholamine receptors may also contribute to haloperidol's mechanism of action. Haloperidol may also inhibit the reuptake of various neurotransmitters in the midbrain, and appears to have a strong central antidopaminergic and weak central anticholinergic activity. The drug produces catalepsy and inhibits spontaneous motor activity and conditioned avoidance behaviours in animals. The exact mechanism of antiemetic action of haloperidol has also not been fully determined, but the drug has been shown to directly affect the chemoreceptor trigger zone (CTZ) through the blocking of dopamine receptors in the CTZ. Toxicity Data LD50: 128 mg/kg (Oral, Rat) LD50: 71 mg/kg (Oral, Mouse) LD50: 90 mg/kg (Oral, Dog) LD50: 165 mg/kg (Oral, Rat) Interactions Prior administration of haloperidol may decrease the pressor response to phenylephrine because of the alpha-adrenergic blocking action of haloperidol. Prior administration of haloperidol may decrease the pressor effect and duration of action of methoxamine because of the alpha-adrenergic blocking action of haloperidol. Concurrent use /of metaraminol/ with haloperidol usually decreases, but does not reverse or completely block, the pressor response to metaraminol, because of the alpha-adrenergic blocking action of haloperidol. Concurrent use /with levodopa or pergolide/ may decrease the therapeutic effects of these agents because of blockade of dopamine receptors by haloperidol. For more Interactions (Complete) data for HALOPERIDOL (26 total), please visit the HSDB record page. Non-Human Toxicity Values LD50 Rat oral 165 mg/kg LD50 Mouse ip 60 mg/kg |
|
| 参考文献 |
|
|
| 其他信息 |
Therapeutic Uses
Anti-Dyskinesia Agents; Antiemetics; Antipsychotic Agents, Butyrophenone; Dopamine Antagonists Haloperidol is indicated for the management of the manifestations of acute and chronic psychotic disorders including schizophrenia, manic states, and drug-induced psychoses, such as steroid psychosis. It may also be useful in the management of aggressive and agitated patients, including patients with organic mental syndrome or mental retardation. Haloperidol decanoate, a long-acting parenteral from, is intended for maintenance use in the management of patients requiring prolonged parenteral therapy, as in chronic schizophrenia. /Included in US product labeling/ Haloperidol is effective in the treatment of children with severe behavior problems of apparently unprovoked, combative, explosive hyperexcitability. It is also effective in the short-term treatment of hyperactivity in children who show excessive motor activity with accompanying conduct disorders such as aggressiveness, impulsiveness, easy frustration, short attention span, and/or rapid mood fluctuations. In these two groups of children, haloperidol should be tried only in patients who fail to respond to psychotherapy or other non-neuroleptic medication. /Included in US product labeling/ Haloperidol is used to control tics and vocalizations of Tourette's syndrome in children and adults. /Included in US product labeling/ For more Therapeutic Uses (Complete) data for HALOPERIDOL (8 total), please visit the HSDB record page. Drug Warnings Pregnancy risk category: C /RISK CANNOT BE RULED OUT. Adequate, well controlled human studies are lacking, and animal studies have shown risk to the fetus or are lacking as well. There is a chance of fetal harm if the drug is given during pregnancy; but the potential benefits may outweigh the potential risk./ Extrapyramidal reactions occur frequently with haloperidol, especially during the first few days of therapy. In most patients, these reactions consist of parkinsonian symptoms (e.g., marked drowsiness and lethargy, drooling or hypersalivation, fixed stare), which are mild to moderate in severity and are usually reversible following discontinuance of the drug. Other adverse neuromuscular reactions have been reported less frequently, but are often more severe, and include feelings of motor restlessness (i.e., akathisia), tardive dystonia, and dystonic reactions (e.g., hyperreflexia, opisthotonos, oculogyric crisis, torticollis, trismus). Possible drowsiness or dizziness; caution when driving, using machinery, or doing things requiring alertness. Possible dizziness or lightheadedness; caution when getting up suddenly from a lying or sitting position. Because of the possibility of transient hypotension and/or precipitation of angina, haloperidol should be used with caution in patients with severe cardiovascular disorders. If hypotension occurs, metaraminol, norepinephrine, or phenylephrine may be used; epinephrine should not be used since haloperidol causes a reversal of epinephrine's vasopressor effects and a further lowering of blood pressure. For more Drug Warnings (Complete) data for HALOPERIDOL (24 total), please visit the HSDB record page. Pharmacodynamics Use of the first-generation antipsychotics (including haloperidol) is considered highly effective for the management of the "positive" symptoms of schizophrenia including hallucinations, hearing voices, aggression/hostility, disorganized speech, and psychomotor agitation. However, this class is limited by the development of movement disorders such as drug-induced parkinsonism, akathisia, dystonia, and tardive dyskinesia, and other side effects including sedation, weight gain, and prolactin changes. Compared to the lower-potency first-generation antipsychotics such as [DB00477], [DB01624], [DB00623], and [DB01403], haloperidol typically demonstrates the least amount of side effects within class, but demonstrates a stronger disposition for causing extrapyramidal symptoms (EPS). Low‐potency medications have a lower affinity for dopamine receptors so that a higher dose is required to effectively treat symptoms of schizophrenia. In addition, they block many receptors other than the primary target (dopamine receptors), such as cholinergic or histaminergic receptors, resulting in a higher incidence of side effects such as sedation, weight gain, and hypotension. The balance between the wanted drug effects on psychotic symptoms and unwanted side effects are largely at play within dopaminergic brain pathways affected by haloperidol. Cortical dopamine-D2-pathways play an important role in regulating these effects and include the nigrostriatal pathway, which is responsible for causing extrapyramidal symptoms (EPS), the mesolimbic and mesocortical pathways, which are responsible for the improvement in positive schizophrenic symptoms, and the tuberoinfundibular dopamine pathway, which is responsible for hyperprolactinemia. A syndrome consisting of potentially irreversible, involuntary, dyskinetic movements may develop in patients. Although the prevalence of the syndrome appears to be highest among the elderly, especially elderly women, it is impossible to rely upon prevalence estimates to predict, at the inception of antipsychotic treatment, which patients are likely to develop the syndrome. Cases of sudden death, QT-prolongation, and Torsades de Pointes have been reported in patients receiving haloperidol. Higher than recommended doses of any formulation and intravenous administration of haloperidol appear to be associated with a higher risk of QT-prolongation and Torsades de Pointes. Although cases have been reported even in the absence of predisposing factors, particular caution is advised in treating patients with other QT-prolonging conditions (including electrolyte imbalance [particularly hypokalemia and hypomagnesemia], drugs known to prolong QT, underlying cardiac abnormalities, hypothyroidism, and familial long QT-syndrome). A potentially fatal symptom complex sometimes referred to as Neuroleptic Malignant Syndrome (NMS) has been reported in association with antipsychotic drugs. Clinical manifestations of NMS are hyperpyrexia, muscle rigidity, altered mental status (including catatonic signs) and evidence of autonomic instability (irregular pulse or blood pressure, tachycardia, diaphoresis, and cardiac dysrhythmias). Additional signs may include elevated creatine phosphokinase, myoglobinuria (rhabdomyolysis) and acute renal failure. |
| 分子式 |
C21H23CLFNO2
|
|---|---|
| 分子量 |
375.86
|
| 精确质量 |
375.14
|
| 元素分析 |
C, 67.11; H, 6.17; Cl, 9.43; F, 5.05; N, 3.73; O, 8.51
|
| CAS号 |
52-86-8
|
| 相关CAS号 |
Haloperidol-d4; 1189986-59-1; Haloperidol-d4-1; 136765-35-0; Haloperidol hydrochloride; 1511-16-6; Haloperidol lactate; 53515-91-6; Haloperidol-d4 N-Oxide; 1246815-56-4; Haloperidol-13C6
|
| PubChem CID |
3559
|
| 外观&性状 |
White to light yellow crystalline powder.
|
| 密度 |
1.2±0.1 g/cm3
|
| 沸点 |
529.0±50.0 °C at 760 mmHg
|
| 熔点 |
152 °C
|
| 闪点 |
273.8±30.1 °C
|
| 蒸汽压 |
0.0±1.5 mmHg at 25°C
|
| 折射率 |
1.581
|
| LogP |
3.01
|
| tPSA |
40.54
|
| 氢键供体(HBD)数目 |
1
|
| 氢键受体(HBA)数目 |
4
|
| 可旋转键数目(RBC) |
6
|
| 重原子数目 |
26
|
| 分子复杂度/Complexity |
451
|
| 定义原子立体中心数目 |
0
|
| SMILES |
ClC1C([H])=C([H])C(=C([H])C=1[H])C1(C([H])([H])C([H])([H])N(C([H])([H])C([H])([H])C([H])([H])C(C2C([H])=C([H])C(=C([H])C=2[H])F)=O)C([H])([H])C1([H])[H])O[H]
|
| InChi Key |
LNEPOXFFQSENCJ-UHFFFAOYSA-N
|
| InChi Code |
InChI=1S/C21H23ClFNO2/c22-18-7-5-17(6-8-18)21(26)11-14-24(15-12-21)13-1-2-20(25)16-3-9-19(23)10-4-16/h3-10,26H,1-2,11-15H2
|
| 化学名 |
4-[4-(4-chlorophenyl)-4-hydroxypiperidin-1-yl]-1-(4-fluorophenyl)butan-1-one
|
| 别名 |
R1625; HSDB3093; R 1625; HSDB 3093; R-1625; HSDB-3093; Eukystol ;Serenace; Haloperidol; Aloperidin; Eukystol; Brotopon; Haldol; Aloperidin; Aloperidol
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
DMSO: ~100 mg/mL (~266.1 mM)
|
|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 1.67 mg/mL (4.44 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 16.7 mg/mL澄清的DMSO储备液加入到400 μL PEG300中,混匀;再向上述溶液中加入50 μL Tween-80,混匀;然后加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: 1.67 mg/mL (4.44 mM) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 悬浊液; 超声助溶。 例如,若需制备1 mL的工作液,可将 100 μL 16.7mg/mL澄清的DMSO储备液加入到900μL 20%SBE-β-CD生理盐水中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 View More
配方 3 中的溶解度: ≥ 1.67 mg/mL (4.44 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.6606 mL | 13.3028 mL | 26.6057 mL | |
| 5 mM | 0.5321 mL | 2.6606 mL | 5.3211 mL | |
| 10 mM | 0.2661 mL | 1.3303 mL | 2.6606 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
| NCT Number | Recruitment | interventions | Conditions | Sponsor/Collaborators | Start Date | Phases |
| NCT03021486 | Active Recruiting |
Other: Chlorpromazine Drug: Haloperidol |
Delirium Advanced Malignant Neoplasm |
M.D. Anderson Cancer Center | June 5, 2017 | Phase 2 Phase 3 |
| NCT01949662 | Active Recruiting |
Drug: Placebo Drug: Haloperidol decanoate |
Advanced Cancers | M.D. Anderson Cancer Center | January 2014 | Phase 2 |
| NCT03392376 | Active Recruiting |
Drug: Haloperidol Injection Other: Saline (0,9%) |
Delirium | Zealand University Hospital | June 13, 2018 | Phase 4 |
| NCT04750395 | Recruiting | Drug: Haloperidol Solution Drug: Olanzapine Tablets |
Delirium Terminal Illness |
HCA Hospice Care | September 1, 2021 | Phase 2 |
| NCT03743649 | Recruiting | Drug: Haloperidol Drug: Lorazepam |
Delirium Metastatic Malignant Neoplasm |
M.D. Anderson Cancer Center | July 17, 2019 | Phase 2 Phase 3 |
 Prosulpride
Prosulpride
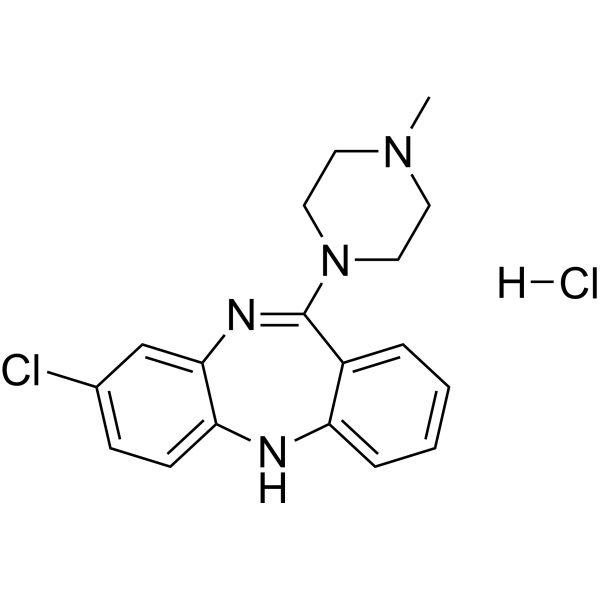 Clozapine hydrochloride
Clozapine hydrochloride
 TAT-D1 peptide
TAT-D1 peptide
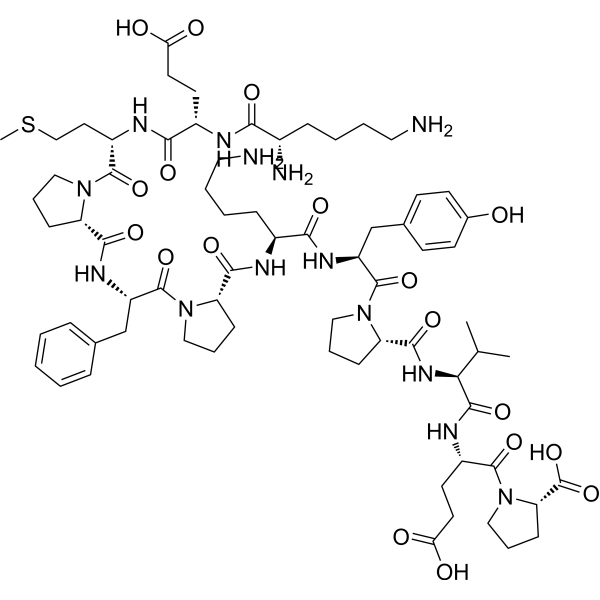 KEMPFPKYPVEP
KEMPFPKYPVEP
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1





























