
| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| 500mg |
|
||
| 1g |
|
||
| Other Sizes |
|
| 靶点 |
PKA (Ki = 48 nM); S6K1 (IC50 = 80 nM); PKG (Ki = 0.48 μM)
|
|---|---|
| 体外研究 (In Vitro) |
当使用 ATP 时,H-89 会竞争性抑制蛋白激酶 A。在 PC12D 细胞中,H-89 以剂量依赖性方式抑制毛喉素诱导的蛋白质磷酸化,而不降低细胞内环 AMP 水平。在 PC12D 细胞中,H-89 强烈抑制毛喉素引起的神经突发育。在 PC12D 细胞裂解物中,H-89 (30 μM) 显着降低 cAMP 依赖性组蛋白 IIb 磷酸化活性 [1]。 H-89 (1-2 μM) 显着延迟了大鼠皮质纤维的重新启动,这很可能是由于它对 T 系统电位产生了负面影响。 H-89 (10-100 μM) 改变大鼠皮肤纤维收缩装置的 Ca32 敏感性,并抑制 SR 的净 Ca2+ 吸收 [2]。
|
| 体内研究 (In Vivo) |
在经过 PTZ 治疗的动物中,H-89(0.2 mg/100g,腹腔注射)显着增加了癫痫潜伏期和阈值。 H-89 显着提高癫痫潜伏期和癫痫阈值,并抑制布克拉地辛 (300 nM) 的致癫痫作用,剂量为 0.05 和 0.2 mg/100 g,腹膜内注射 [3]。
H-89预处理对PTZ诱发癫痫的影响[3] 图2A和B显示了用不同剂量的H-89(0.05、0.1和0.2 mg/100 g,i.p.,30分钟)预处理对PTZ(0.5%w/v i.v)诱导的癫痫发作的影响。与对照组相比,以0.2 mg/100克的剂量给药H-89显著增加了癫痫发作的潜伏期和阈值(***p<0.001)。与对照组动物相比,其他两种剂量的H-89(0.05和0.1 mg/100 g)在癫痫发作潜伏期和阈值方面没有观察到显著差异(图2A和B)。 己酮可可碱和H-89联合预处理对PTZ诱导的小鼠癫痫发作的影响[3] 属于该组合组的所有动物在PTZ输注前45分钟接受PTX作为第一组分,30分钟接受H-89作为第二组分。与对照组相比,接受PTX 50 mg/kg和H-89 0.2 mg/kg以及PTX 100 mg/kg和H-890.2 mg/100 g的组在癫痫发作潜伏期和阈值方面存在显著差异(***P<0.001)(图4A和B)。PTX(50和100 mg/kg)给药显著减弱了H-89(0.2 mg/100 g)对癫痫发作阈值和潜伏期的影响(*P<0.05)(图4A和B)。 |
| 酶活实验 |
cAMP 依赖性蛋白激酶活性在最终体积为 0.2 mL 的反应混合物中进行测定,其中包含 50 mM Tris-HCl (pH 7.0)、10 mM 醋酸镁、2 mM EGTA、1 μM cAMP 或不含 cAMP,3.3 -20 μM [γ-32P]ATP (4 × 105 cpm)、0.5 μg 酶、100 μg 组蛋白 H2B 和每种化合物,如所示。
|
| 细胞实验 |
测定细胞内cAMP 的水平。培养 48 小时后,PC12D 细胞在含有 30 μM H-89 的测试培养基中生长 1 小时,然后暴露于含有 10 μM 毛喉素和 30 μM H-89 的全新培养基中。添加 0.5 ml 6% 三氯乙酸,同时用橡胶警察刮下细胞并进行超声处理。加入2ml石油醚,混合,3000rpm离心10分钟,提取三氯乙酸。吸出顶层后,残留样品溶液用于分析。
|
| 动物实验 |
rat; mice
20 or 200 mg/kg (Rat); 0-5 mg/kg (Mice) s.c. (Rat); i.p. (Mice) Pentoxifylline (25, 50, 100 mg/kg), bucladesine (50, 100, 300 nM/mouse) and H-89 (0.05, 0.1, 0.2 mg/100 g) were administered intraperitoneally (i.p.) 30 min before intravenous (i.v.) infusion of PTZ. In combination groups, the first and second components were injected 45 and 30 min before PTZ infusion. In all groups, the respective control animals received an appropriate volume of vehicle. For the i.v. infusion, the needle was inserted into the lateral tail vein, fixed to the tail vein by a narrow piece of adhesive tape, and the animal was allowed to move freely (Gholipour et al., 2008, 2009). PTZ solution was infused at a concentration rate of 1 ml/min.[3] |
| 参考文献 |
|
| 其他信息 |
N-[2-(4-bromocinnamylamino)ethyl]isoquinoline-5-sulfonamide is a member of the class of isoquinolines that is the sulfonamide obtained by formal condensation of the sulfo group of isoquinoline-5-sulfonic acid with the primary amino group of N(1)-[3-(4-bromophenyl)prop-2-en-1-yl]ethane-1,2-diamine. It is a protein kinase A inhibitor. It has a role as an EC 2.7.11.11 (cAMP-dependent protein kinase) inhibitor. It is a member of isoquinolines, a sulfonamide, a member of bromobenzenes, an olefinic compound and a secondary amino compound. It is a conjugate base of a N-[2-(4-bromocinnamylamino)ethyl]isoquinoline-5-sulfonamide(2+).
A newly synthesized isoquinolinesulfonamide, H-89 (N-[2-(p-bromocinnamylamino)ethyl]-5-isoquinoline-sulfonamide), was shown to have a potent and selective inhibitory action against cyclic AMP-dependent protein kinase (protein kinase A), with an inhibition constant of 0.048 +/- 0.008 microM. H-89 exhibited weak inhibitory action against other kinases and Ki values of the compound for these kinases, including cGMP-dependent protein kinase (protein kinase G), Ca2+/phospholipid-dependent protein kinase (protein kinase C), casein kinase I and II, myosin light chain kinase, and Ca2+/calmodulin-dependent protein kinase II were 0.48 +/- 0.13, 31.7 +/- 15.9, 38.3 +/- 6.0, 136.7 +/- 17.0, 28.3 +/- 17.5, and 29.7 +/- 8.1 microM, respectively. Kinetic analysis indicated that H-89 inhibits protein kinase A, in competitive fashion against ATP. To examine the role of protein kinase A in neurite outgrowth of PC12 cells, H-89 was applied along with nerve growth factor (NGF), forskolin, or dibutyryl cAMP. Pretreatment with H-89 led to a dose-dependent inhibition of the forskolin-induced protein phosphorylation, with no decrease in intracellular cyclic AMP levels in PC12D cells, and the NGF-induced protein phosphorylation was not not inhibited. H-89 also significantly inhibited the forskolin-induced neurite outgrowth from PC12D cells. This inhibition also occurred when H-89 was added before the addition of dibutyryl cAMP. Pretreatment of PC12D cells with H-89 (30 microM) inhibited significantly cAMP-dependent histone IIb phosphorylation activity in cell lysates but did not affect other protein phosphorylation activity such as cGMP-dependent histone IIb phosphorylation activity, Ca2+/phospholipid-dependent histone IIIs phosphorylation activity, Ca2+/calmodulin-dependent myosin light chain phosphorylation activity, and alpha-casein phosphorylation activity. However, this protein kinase A inhibitor did not inhibit the NGF-induced neurite outgrowth from PC12D cells. Thus, the forskolin- and dibutyryl cAMP-induced neurite outgrowth is apparently mediated by protein kinase A while the NGF-induced neurite outgrowth is mediated by a protein kinase A-independent pathway.[1] The specificities of 28 commercially available compounds reported to be relatively selective inhibitors of particular serine/threonine-specific protein kinases have been examined against a large panel of protein kinases. The compounds KT 5720, Rottlerin and quercetin were found to inhibit many protein kinases, sometimes much more potently than their presumed targets, and conclusions drawn from their use in cell-based experiments are likely to be erroneous. Ro 318220 and related bisindoylmaleimides, as well as H89, HA1077 and Y 27632, were more selective inhibitors, but still inhibited two or more protein kinases with similar potency. LY 294002 was found to inhibit casein kinase-2 with similar potency to phosphoinositide (phosphatidylinositol) 3-kinase. The compounds with the most impressive selectivity profiles were KN62, PD 98059, U0126, PD 184352, rapamycin, wortmannin, SB 203580 and SB 202190. U0126 and PD 184352, like PD 98059, were found to block the mitogen-activated protein kinase (MAPK) cascade in cell-based assays by preventing the activation of MAPK kinase (MKK1), and not by inhibiting MKK1 activity directly. Apart from rapamycin and PD 184352, even the most selective inhibitors affected at least one additional protein kinase. Our results demonstrate that the specificities of protein kinase inhibitors cannot be assessed simply by studying their effect on kinases that are closely related in primary structure. We propose guidelines for the use of protein kinase inhibitors in cell-based assays.[2] H89 is marketed as a selective and potent inhibitor of protein kinase A (PKA). Since its discovery, it has been used extensively for evaluation of the role of PKA in the heart, osteoblasts, hepatocytes, smooth muscle cells, neuronal tissue, epithelial cells, etc. Despite the frequent use of H89, its mode of specific inhibition of PKA is still not completely understood. It has also been shown that H89 inhibits at least 8 other kinases, while having a relatively large number of PKA-independent effects which may seriously compromise interpretation of data. Thus, while recognizing its kinase inhibiting properties, it is advised that H89 should not be used as the single source of evidence of PKA involvement. H-89 should be used in conjunction with other PKA inhibitors, such as Rp-cAMPS or PKA analogs.[3] |
| 分子式 |
C20H20BRN3O2S
|
|---|---|
| 分子量 |
446.363
|
| 精确质量 |
445.045
|
| 元素分析 |
C, 53.82; H, 4.52; Br, 17.90; N, 9.41; O, 7.17; S, 7.18
|
| CAS号 |
127243-85-0
|
| 相关CAS号 |
H-89 dihydrochloride;130964-39-5; 127243-85-0 ; 1000995-75-4; 1049740-55-7 (2HCl hydrate)
|
| PubChem CID |
449241
|
| 外观&性状 |
Typically exists as off-white to light brown solids at room temperature
|
| 密度 |
1.4±0.1 g/cm3
|
| 沸点 |
639.7±65.0 °C at 760 mmHg
|
| 熔点 |
195-200°C
|
| 闪点 |
340.7±34.3 °C
|
| 蒸汽压 |
0.0±1.9 mmHg at 25°C
|
| 折射率 |
1.653
|
| LogP |
3.03
|
| tPSA |
79.47
|
| 氢键供体(HBD)数目 |
2
|
| 氢键受体(HBA)数目 |
5
|
| 可旋转键数目(RBC) |
8
|
| 重原子数目 |
27
|
| 分子复杂度/Complexity |
570
|
| 定义原子立体中心数目 |
0
|
| SMILES |
BrC1C([H])=C([H])C(=C([H])C=1[H])/C(/[H])=C(\[H])/C([H])([H])N([H])C([H])([H])C([H])([H])N([H])S(C1=C([H])C([H])=C([H])C2C([H])=NC([H])=C([H])C1=2)(=O)=O
|
| InChi Key |
N-[2-[[3-(4-Bromophenyl)-2-propen-1-yl]amino]ethyl]-5-Isoquinolinesulfonamide
|
| InChi Code |
ZKZXNDJNWUTGDK-NSCUHMNNSA-N
|
| 化学名 |
H-89 free base H-89 free base H 89 H89.
|
| 别名 |
h-89; 127243-85-0; H 89; N-(2-(4-Bromocinnamylamino)ethyl)-5-isoquinolinesulfonamide; H-89 DIHYDROCHLORIDE; N-[2-[[(E)-3-(4-bromophenyl)prop-2-enyl]amino]ethyl]isoquinoline-5-sulfonamide; Protein kinase inhibitor H-89; N-[2-(P-BROMOCINNAMYLAMINO)ETHYL]-5-ISOQUINOLINE SULFONAMIDE;
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
DMSO : ~100 mg/mL (~224.03 mM)
|
|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 2.5 mg/mL (5.60 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 25.0 mg/mL澄清DMSO储备液加入到400 μL PEG300中,混匀;然后向上述溶液中加入50 μL Tween-80,混匀;加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: 2.5 mg/mL (5.60 mM) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 悬浊液; 超声助溶。 例如,若需制备1 mL的工作液,可将 100 μL 25.0 mg/mL澄清DMSO储备液加入900 μL 20% SBE-β-CD生理盐水溶液中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.2403 mL | 11.2017 mL | 22.4034 mL | |
| 5 mM | 0.4481 mL | 2.2403 mL | 4.4807 mL | |
| 10 mM | 0.2240 mL | 1.1202 mL | 2.2403 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
 Kiss2 peptide acetate
Kiss2 peptide acetate
 Rp-8-Br-cAMPS sodium
Rp-8-Br-cAMPS sodium
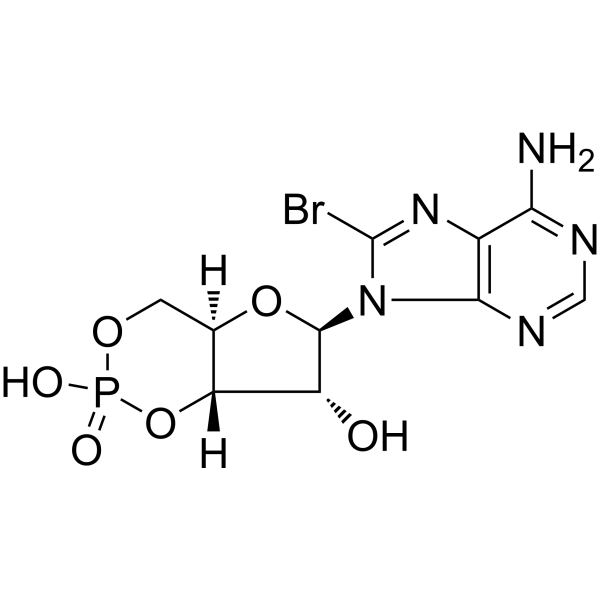 8-Bromo-cAMP
8-Bromo-cAMP
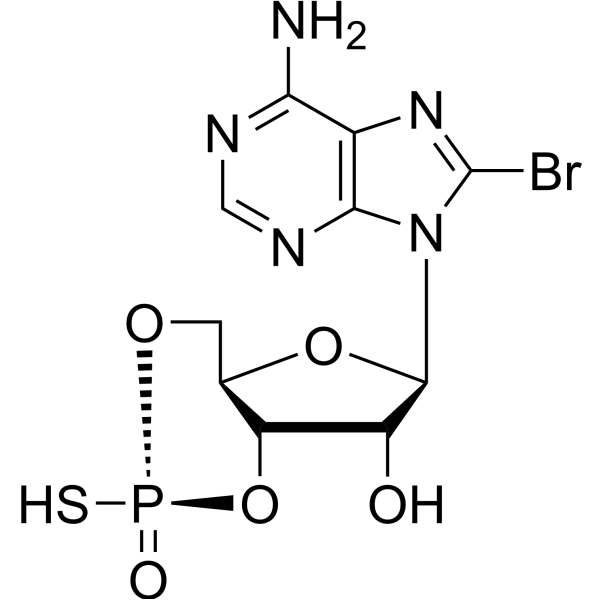 Sp-8-Br-cAMPS
Sp-8-Br-cAMPS
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1






























 COA
COA
