| 规格 | 价格 | |
|---|---|---|
| 500mg | ||
| 1g | ||
| Other Sizes |
| 靶点 |
PPARα
|
|---|---|
| 体外研究 (In Vitro) |
过氧化物酶体增殖物激活受体PPARα、PPARγ和PPARδ是配体激活的转录因子,在脂质稳态中起着关键作用。贝特类药物提高了高密度脂蛋白胆固醇的循环水平,降低了甘油三酯的水平,部分原因是它们作为PPARalpha激动剂的活性;然而,贝特类药物的低效力和有限的选择性可能会限制其疗效,因此需要开发更有效和更具选择性的PPARalpha激动剂。选择性PPARdelta激动剂1(GW501516)的修饰,以掺入贝特类的2-芳基-2-甲基丙酸基团,导致效力和选择性明显向PPARα激动作用转变。该系列的优化得到25a,其显示PPARalpha的EC50=4nM,与PPARdelta和PPARgamma相比,选择性至少为500倍。化合物25a(GW590735)已进入治疗脂质失衡疾病的临床试验[1]。
|
| 体内研究 (In Vivo) |
GW 590735(0.5–5 mg/kg;口服,每日两次,持续 5 天)可在 Apo-AI 转基因小鼠模型(人类转基因雄性 C57BL/6 小鼠)中升高 HDL 胆固醇并降低 LDLc 和 TG。 APOA-II)(1)。 2.4 小时后,分别显示 GW 590735(IV;2.7 mg/kg;大鼠)的 Cl、Vd、T1/2 和 F%、1 L/kg、5 mL/min/kg 和 47% [1] 。 GW 590735(IV;2 mg/kg;狗)治疗结果显示,Cl、Vd、T1/2 和 F% 分别为 13 mL/min/kg、2.8 L/kg、2.6 小时和85% [1]。
|
| 酶活实验 |
然后用缓冲液C将蛋白质稀释至1mg/mL,使最终缓冲液组合物为220mM乙酸铵、20mM HEPES pH 7.5、1mM EDTA和1mM DTT。肽SRC116以1.5的摩尔比作为2mg/100μL DMSO储备加入。然后以5:1的摩尔比加入2 mg/100μL DMSO原液中的配体,并在4 K下旋转20分钟以澄清溶液,然后在Centriprep 10过滤装置中浓缩。将含有PPARαLBD-SRC1复合物的溶液浓缩至约10mg/mL,产率为80%[1]。
|
| 细胞实验 |
使用质粒载体pRSETA的T7启动子表达具有N端6xHis标签的PPARα配体结合结构域(氨基酸192−468)。用该表达载体转化的BL21(DE3)大肠杆菌细胞在24°C的摇瓶中生长66小时。收获细胞,重新悬浮并裂解。将裂解的细胞离心,并将上清液装载在镍琼脂糖柱上。用150mL缓冲液A(10%甘油,20mM HEPES pH 7.5,25mM咪唑)洗涤柱,用450mL缓冲液B梯度(10%甘油、20mM HEPES pH 7.5,500mM咪唑)洗脱蛋白质。在20%缓冲液B中洗脱的蛋白质用一倍体积的缓冲液C(20mM HEPES,pH 7.5,1mM EDTA)稀释,并装载在100mL S-Sepharose柱上。用100mL缓冲液C洗涤柱,用200mL梯度缓冲液D(20mM HEPES,pH 7.5,10mM DTT,1M乙酸铵)洗脱PPARαLBD蛋白。PPARαLBD在43%缓冲液D下从柱中洗脱。SDS-PAGE分析确定,蛋白质产量为9mg/L生长的细胞,纯度>95%[1]。
|
| 动物实验 |
Pharmacokinetics in Rat and Dog.[1]
Compound 25a was administered to Wistar rats (n = 15) by oral gavage at dose of 3 mg/kg in pH 7 buffer, 0.1% Tween80 and by intravenous injection via the penis vein (n = 30) at a dose of 2.7 mg/kg in 10% DMSO and PEG200. Compound 25a was administered orally to male beagle dogs (n = 3) by stomach intubation at a dose of 3 mg/kg in pH 7 buffer, 0.1% Tween80 and by intravenous injection via the cephalic vein at a dose of 2 mg/kg in 10% DMSO and PEG200. Blood samples were placed on wet ice, and plasma was collected after centrifugation. Plasma samples were stored frozen at −20 °C until time of analysis. Plasma samples (0.5 mL) were diluted with 1:1 buffer (NaH2PO4, 0.1 M, pH 4) and then extracted with ethyl acetate (5 mL). The ethyl acetate was evaporated, and the residue was resuspended in 200 μL of mobile phase (water/acetonitrile/TFA; 30v/70v/0.1%). Samples were analyzed by high-performance liquid chromatography spectrometric analysis (LC/MS/MS). Pharmacokinetic parameters were determined by SIPHAR. Apo-A-I Transgenic Mouse Model. [1] Male C57BL/6 mice transgenic for human ApoA-I were randomized into treatment groups of n = 5 animals. Twice a day oral administration of vehicle (0.5% HPMC/1% Tween80, pH = 7.0) or indicated doses of compound as a suspension began when animals were nine weeks old and lasted for 5 days. Animals were fasted overnight before blood samples were taken by intracardiac puncture. Whole liver was collected and weighed. Blood samples were left for 30 min at 37 °C to coagulate and centrifuged 10 min at 10 000 rpm. Total serum fraction was then collected and frozen at −20 °C until use. Total cholesterol and total TG were dosed using kits 61219 and 61236, respectively, following manufacturer instructions. After 10 min of incubation at 37 °C, the colorimetric reaction was read at 492 nm with an iEMS reader. Cholesterol HDL, LDL, and VLDL fractions were separated by HPLC. Samples were diluted 1/5 in phosphate buffer (Ca++ and Mg- free) and filtered on 0.45 μm to remove excess proteins before HPLC. All changes reported with an asterisk are statistically significant (p < 0.05) as determined by one-way ANOVA analysis. |
| 药代性质 (ADME/PK) |
Pharmacokinetics in Rat and Dog.[1]
Following intravenous administration of Compound 25a (GW590735) (2.7 mg/kg) to the rat, distribution to the tissues was limited with the volume of distribution (1 L/kg) similar to that of total body water (0.6 L/kg). Total plasma clearance was low (5 mL/min/kg), representing about 6% of rat hepatic blood flow. The low clearance and moderate volume of distribution resulted in a plasma half-life of 2.4 h. Following a single oral dose of compound 25a at 3 mg/kg, the maximum concentration of compound in the plasma was 1461 ng/mL after 1.5 h. The bioavailability was high (47%). Following intravenous administration of compound 25a (GW590735) to the dog at 2 mg/kg, distribution to the tissues was limited with the volume of distribution (2.8 L/kg) being greater than that of total body water (0.6 L/kg). Total plasma clearance was moderate (13 mL/min/kg), representing about 35% of hepatic blood flow in the dog. The moderate clearance and low volume of distribution resulted in a plasma half-life of 2.6 h. Following a single oral dose of compound 25a at 3 mg/kg, the maximum concentration of compound in the plasma was 1449 ng/mL. The bioavailability was high (85%). |
| 参考文献 | |
| 其他信息 |
Pharmacokinetics in Rat and Dog.
Following intravenous administration of compound 25a (2.7 mg/kg) to the rat, distribution to the tissues was limited with the volume of distribution (1 L/kg) similar to that of total body water (0.6 L/kg). Total plasma clearance was low (5 mL/min/kg), representing about 6% of rat hepatic blood flow. The low clearance and moderate volume of distribution resulted in a plasma half-life of 2.4 h. Following a single oral dose of compound 25a at 3 mg/kg, the maximum concentration of compound in the plasma was 1461 ng/mL after 1.5 h. The bioavailability was high (47%). Following intravenous administration of compound 25a to the dog at 2 mg/kg, distribution to the tissues was limited with the volume of distribution (2.8 L/kg) being greater than that of total body water (0.6 L/kg). Total plasma clearance was moderate (13 mL/min/kg), representing about 35% of hepatic blood flow in the dog. The moderate clearance and low volume of distribution resulted in a plasma half-life of 2.6 h. Following a single oral dose of compound 25a at 3 mg/kg, the maximum concentration of compound in the plasma was 1449 ng/mL. The bioavailability was high (85%).[1] In Vivo Pharmacology. Several compounds in this series demonstrated profound in vivo activity in animal models of dyslipidemia, as illustrated by compound 25a. The human Apo-A-I-transgenic mouse model has been proposed to be potentially relevant to human disease, because in this model, fibrates give upregulation rather than the repression of Apo-A-I seen in other rodent models. Compound 25a shows similar PPARα agonist potency and selectivity versus murine and human PPARs (murine PPAR EC50, α = 15 nM; δ = 1000 nM; γ > 10 000 nM). When administered orally twice a day for 5 days, 25a, prepared as a suspension in 0.5% HPMC 100/1% Tween80 at pH = 7.0, gave dose-related decreases in circulating TG, VLDLc, and LDLc and concomitant increases in HDLc (Table 7). The ED50 for the HDL effect was approximately 1 mg/kg. Circulating levels of Apo-A-I were also increased by the treatment (data not shown), consistent with the mechanism of action. As a reference, Fenofibrate was tested in human Apo-A-I transgenic mice at 50 mg/kg. It produced the expected profile with a decrease in plasma TG (−43%), VLDL cholesterol (−64%), and LDL cholesterol (−78%), as well as an increase in HDL cholesterol (+26%). The finding that 25a is able to lower LDLc and TG and increase HDL cholesterol in the Apo-A-I-transgenic mouse model suggests that it will deliver significant therapeutic benefit in the treatment of dyslipidemia and hypertriglyceridemia. Compound 25a (GW590735) has been progressed to clinical trials for the treatment of diseases of lipid imbalance.[1] Starting from the selective PPARδ agonist 1, which showed weak potency on PPARα, we have developed a new series of highly potent and selective PPARα agonists, one of which, 25a, has progressed to clinical evaluation. The superior potency and PPAR subtype selectivity of 25a suggest that this compound offers the potential to deliver significantly improved therapeutic benefits over the fibrates in dyslipidemia and hypertriglyceridemia. |
| 精确质量 |
478.12
|
|---|---|
| 元素分析 |
C, 55.20; H, 4.03; F, 11.39; N, 5.60; Na, 4.59; O, 12.79; S, 6.41
|
| CAS号 |
343322-50-9
|
| 相关CAS号 |
343322-50-9 (sodium); 343321-96-0; 622402-22-6 (free acid);
|
| 外观&性状 |
Typically exists as solid at room temperature
|
| LogP |
5.22
|
| InChi Key |
RKWLGVFEDFICPQ-UHFFFAOYSA-M
|
| InChi Code |
InChI=1S/C23H21F3N2O4S.Na/c1-13-18(33-20(28-13)15-6-8-16(9-7-15)23(24,25)26)19(29)27-12-14-4-10-17(11-5-14)32-22(2,3)21(30)31;/h4-11H,12H2,1-3H3,(H,27,29)(H,30,31);/q;+1/p-1
|
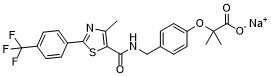
| 化学名 |
Propanoic acid, 2-methyl-2-(4-((((4-methyl-2-(4-(trifluoromethyl)phenyl)-5-thiazolyl)carbonyl)amino)methyl)phenoxy)- sodium
|
| 别名 |
GW 590735 sodium; GW-590735 sodium;343321-96-0; 622402-22-6; GW590735; GW-590735; 2-methyl-2-(4-((4-methyl-2-(4-(trifluoromethyl)phenyl)thiazole-5-carboxamido)methyl)phenoxy)propanoic acid; QKY617BBX5; CHEMBL219586; GW590735 sodium
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples
|
|---|---|
| 溶解度 (体内实验) |
注意: 如下所列的是一些常用的体内动物实验溶解配方,主要用于溶解难溶或不溶于水的产品(水溶度<1 mg/mL)。 建议您先取少量样品进行尝试,如该配方可行,再根据实验需求增加样品量。
注射用配方
注射用配方1: DMSO : Tween 80: Saline = 10 : 5 : 85 (如: 100 μL DMSO → 50 μL Tween 80 → 850 μL Saline)(IP/IV/IM/SC等) *生理盐水/Saline的制备:将0.9g氯化钠/NaCl溶解在100 mL ddH ₂ O中,得到澄清溶液。 注射用配方 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (如: 100 μL DMSO → 400 μL PEG300 → 50 μL Tween 80 → 450 μL Saline) 注射用配方 3: DMSO : Corn oil = 10 : 90 (如: 100 μL DMSO → 900 μL Corn oil) 示例: 以注射用配方 3 (DMSO : Corn oil = 10 : 90) 为例说明, 如果要配制 1 mL 2.5 mg/mL的工作液, 您可以取 100 μL 25 mg/mL 澄清的 DMSO 储备液,加到 900 μL Corn oil/玉米油中, 混合均匀。 View More
注射用配方 4: DMSO : 20% SBE-β-CD in Saline = 10 : 90 [如:100 μL DMSO → 900 μL (20% SBE-β-CD in Saline)] 口服配方
口服配方 1: 悬浮于0.5% CMC Na (羧甲基纤维素钠) 口服配方 2: 悬浮于0.5% Carboxymethyl cellulose (羧甲基纤维素) 示例: 以口服配方 1 (悬浮于 0.5% CMC Na)为例说明, 如果要配制 100 mL 2.5 mg/mL 的工作液, 您可以先取0.5g CMC Na并将其溶解于100mL ddH2O中,得到0.5%CMC-Na澄清溶液;然后将250 mg待测化合物加到100 mL前述 0.5%CMC Na溶液中,得到悬浮液。 View More
口服配方 3: 溶解于 PEG400 (聚乙二醇400) 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
| NCT Number | Recruitment | interventions | Conditions | Sponsor/Collaborators | Start Date | Phases |
| NCT00169559 | Completed | Drug: GW590735 Drug: fenofibrate |
Dyslipidaemias | GlaxoSmithKline | November 2003 | Phase 2 |
| NCT00388180 | Completed | Drug: GW501516 Drug: GW590735 |
Obesity Dyslipidaemias |
GlaxoSmithKline | December 2004 |
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1


































