| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 500mg |
|
||
| 1g |
|
||
| 5g |
|
||
| Other Sizes |
|

| 靶点 |
Topoisomerase II (IC50 = 36.7 μM)
|
|---|---|
| 体外研究 (In Vitro) |
盐酸加替沙星靶向金黄色葡萄球菌 MS5935 拓扑异构酶 IV、大肠杆菌 NIHJ JC-2 DNA 旋转酶和 HeLa 细胞拓扑异构酶 II,IC50 值分别为 13.8 μg/ml 和 0.109 μg/ml。和 265 μg/ml [1]。盐酸加替沙星靶向金黄色葡萄球菌 MS5935 拓扑异构酶 IV、大肠杆菌 NIHJ JC-2 DNA 旋转酶和 HeLa 细胞拓扑异构酶 II,MIC 值分别为 0.05 μg/ml、0.0063 μg/ml 和 122。微克/毫升[1]。盐酸加替沙星对野生型菌株(MS5935、MS5952、MR5867 和 MR6009)、步骤一、步骤二、步骤三和步骤四突变体表现出抗菌活性,MIC 值为 0.05 至 0.10 μg/ml、0.20 μg/ml、1.56 至分别为3.13μg/ml、1.56至6.25μg/ml和50至200μg/ml。除MS5935菌株的第二步突变体外,盐酸加替沙星对第二步和第三步突变体(MS5952、MR5867和MR6009)表现出最强的功效[2]。加替沙星盐酸盐对norA转化体NY12具有有效的活性(MIC,0.39 μg/ml)[2]。盐酸加替沙星(20-100 μM;72 小时)在第 1 天显着降低胰岛素含量至 60%,并在第 3 天继续通过 20 μM 和 100 μM 盐酸加替沙星分别降低至 50.1% 和 50.1%。 44.7%[3]。
|
| 体内研究 (In Vivo) |
在感染巴西诺卡氏菌的小鼠中,盐酸加替沙星(皮下注射;100 mg/kg;每天3次;30天)显着降低了足垫病变的频率[4]。
加替沙星可降低正常和糖尿病大鼠的血清葡萄糖浓度。加替沙星导致正常和糖尿病大鼠血清肾上腺素浓度增加。
加替沙星皮下注射;100毫克/公斤;每日三次;30 d)可显著减少巴西诺卡菌小鼠脚垫损伤数[4]
100 mg/kg的加替沙星使巴西奈瑟菌HUJEG-1的血药浓度高于MIC (0.25 μg/ml) 4小时以上,血清中最高浓度为18 μg/ml(图1)。25 mg/kg的利奈唑胺也使其血药浓度高于MIC (0.12 μg/ml) 4小时以上,血清中最高浓度为50 μg/ml。考虑到这些结果,我们决定使用加替沙星100mg /kg,每日3次,皮下注射,利奈唑胺25mg /kg,每日3次。图2显示了加替沙星对病变发展的影响。这些动物显示出病变数量的减少,与利奈唑胺的效果相当。单因素方差分析结果显示,与生理盐水组相比,利奈唑胺组和加替沙星组均有统计学意义,P值为0.001。[4] 加替沙星停药对胰岛素分泌及胰岛胰岛素含量的影响[3] 小鼠胰岛在20 μM或100 μM加替沙星中培养1天,用不含加替沙星的RPMI培养基彻底清洗,再在不含加替沙星的培养基中培养2天。加替沙星使葡萄糖诱导的胰岛素分泌大大减少,但在培养基中去除加替沙星后恢复(图5A)。加替沙星同样降低了胰岛胰岛素含量,在20 μM加替沙星组中(为对照组(0 μM加替沙星)的77%),在100 μM加替沙星组中,胰岛胰岛素含量几乎没有恢复(图5B)。由于加替沙星的存在降低了胰岛胰岛素含量,因此胰岛素分泌量以%表示,20 μM加替沙星和100 μM加替沙星培养的胰岛胰岛素释放在停药后几乎完全恢复(图5C)。 |
| 酶活实验 |
细菌酶 DNA 旋转酶和拓扑异构酶 IV 受到抗生素加替沙星的抑制,加替沙星属于第四代氟喹诺酮家族。
酶试验。[1] 用Peng和Marians的方法测定了重组的拓扑异构酶IV的十烷癸烯化活性,并进行了少量修改。电泳分析反应,溴化乙啶染色后琼脂糖凝胶中DNA定量。用fmbioii Multi-View密度分析仪测定了着丝体DNA十烯二化单体对应的条带亮度。 DNA旋切酶的超旋切活性由Gellert等人的方法测定,稍作修改。按照拓扑异构酶IV测定方法进行分析。 拓扑异构酶II的松弛活性采用Oomori等的方法测定。 通过测定抑制50%酶反应所需的浓度(IC50)来测定每种喹诺酮类药物对II型拓扑异构酶的抑制作用。通过将HeLa细胞拓扑异构酶II的IC50除以细菌II型拓扑异构酶的IC50来确定选择性。 |
| 细胞实验 |
抗菌 与第四代氟喹诺酮抗生素家族的其他成员一样,加替沙星抑制细菌酶 DNA 旋转酶和拓扑异构酶 IV。当涉及第二步突变体(grlA gyrA;加替沙星 MIC 范围,1.56 至 3.13 microg/ml)和第三步突变体(grlA gyrA grlA;加替沙星 MIC 范围,1.56 至 6.25 microg/ml)时,加替沙星表现出活性与托舒沙星相当,比诺氟沙星、氧氟沙星、环丙沙星和司帕沙星更有效。这些结果表明,在所研究的喹诺酮类药物中,加替沙星对单突变拓扑 IV 和单突变 DNA 旋转酶具有最有效的抑制活性。对于铜绿假单胞菌感染的角膜溃疡,眼用 0.3% 加替沙星在给药频率较低时至少与环丙沙星一样有效。荧光素保留分数显示出有利于加替沙星的趋势。
小鼠胰岛和MIN6细胞的胰岛素-2 mRNA分析[3] 50组胰岛在加或不加gatifloxacin培养3天后,使用poly(A) Pure试剂盒分离poly(A)+ rna,根据制造商说明使用SuperScript™II逆转录酶系统合成第一链cdna。TaqMan™小鼠胰岛素-2 (min -2)定量聚合酶链反应(PCR)检测在ABI PRISM™7000序列检测系统中使用正向和反向min -2特异性引物和探针。结果用min -2 mRNA与小鼠甘油醛-3-磷酸脱氢酶(GAPDH) mRNA的比值表示。 MIN6细胞在添加25 mM葡萄糖和13%胎牛血清(含或不含gatifloxacin)的Dulbecco's Minimal Essential培养基中培养3天。用TRIzol试剂制备的总RNA (10 μg)进行Northern blot分析。小鼠β-肌动蛋白mRNA进行标准化。 使用双荧光素酶报告基因检测系统(double -luciferase reporter Assay System),根据生产商说明,在转染人胰岛素启动子-荧光素酶报告基因的MIN6细胞中,用100 μM加替沙星或不加加替沙星培养3天,评估胰岛素启动子活性。相对于加替沙星未处理对照,荧光素酶活性的平均值从重复孔计算。 |
| 动物实验 |
Animal/Disease Models: Female balb/c (Bagg ALBino) mouse, infected with Nocardia brasiliensis on the right hind footpad [1]
Doses: 100 mg/kg Route of Administration: subcutaneous injection; 3 times a day; 30-day Experimental Results: diminished mouse injuries of production. Isolation of mouse pancreatic islets and islet culture [3] Pancreatic islets were isolated from fed male C57Bl/6 mice aged 12–16 weeks by collagenase digestion method. For short term exposure, fresh islets were used. For long term exposure, islets were cultured with or without gatifloxacin in RPMI medium containing 10% fetal bovine serum and 11.1 mM glucose, and used after the indicated culture periods for subsequent experiments. In some experiments (Fig. 5), the islets were cultured in the presence of 20 or 100 μM gatifloxacin for one day, washed with gatifloxacin-free RPMI medium three times to remove remaining gatifloxacin in the culture medium, and then cultured for additional two days in gatifloxacin-free medium. For the animal assays, we utilized Nocardia brasiliensis HUJEG-1, which has been utilized in previous studies. The MICs of this strain, determined by the broth microdilution method, are 0.25 μg/ml for gatifloxacin and 0.12 μg/ml for linezolid. For the determination of the plasma levels of gatifloxacin and linezolid, several doses of these compounds were used. Linezolid was used at 10 mg/kg body weight, 25 mg/kg, and 50 mg/kg, and gatifloxacin at 50 mg/kg, 75 mg/kg, and 100 mg/kg. Eight- to 12-week-old female BALB/c mice were injected subcutaneously with the antimicrobials. For each dose tested, 27 mice were utilized; 24 were injected with the selected dose, and 3 mice were not injected to represent time zero. Next, 500-μl blood samples were taken from the infraorbital sinus of each mouse, which previously had undergone general anesthesia with ethylic ether. The samples were taken from groups of three mice each at the following time intervals: 0 min, 20 min, 40 min, 1 h, 2 h, 4 h, 6 h, 8 h, and 10 h. After sample collection, the plastic tubes containing the blood were centrifuged and the plasma was separated and frozen at −70°C. Plasma concentrations were determined by using a previously validated high performance liquid chromatography method. For the therapeutic assays, 8- to 12-week-old female BALB/c mice were inoculated with 20 mg of Nocardia brasiliensis in the right hind footpad. Seven days later, the therapeutic assay was started. Groups of 15 mice each were used. One group was injected subcutaneously in the back with 0.1 ml of pyrogen-free saline; the rest were treated with either gatifloxacin at 100 mg/kg or linezolid at 25 mg/kg. All the treatments, including the saline solution, were given subcutaneously on the back three times per day during a 4-week period. The development of lesions in the footpad of the animals was scored by two independent readers as described previously. This system classifies the lesions from those animals presenting absolutely no lesions or inflammation as negative or zero and the lesions from animals presenting severe lesions extending above the metatarsal bones as 4+. Differences among the therapeutic groups were analyzed using the analysis of variance test and confirmed with the Dunnet analysis. [4] |
| 药代性质 (ADME/PK) |
Absorption, Distribution and Excretion
Well absorbed from the gastrointestinal tract after oral administration with absolute bioavailability of gatifloxacin is 96% Metabolism / Metabolites Gatifloxacin undergoes limited biotransformation in humans with less than 1% of the dose excreted in the urine as ethylenediamine and methylethylenediamine metabolites Gatifloxacin undergoes limited biotransformation in humans with less than 1% of the dose excreted in the urine as ethylenediamine and methylethylenediamine metabolites Half Life: 7-14 hours Biological Half-Life 7-14 hours |
| 毒性/毒理 (Toxicokinetics/TK) |
Toxicity Summary
The bactericidal action of Gatifloxacin results from inhibition of the enzymes topoisomerase II (DNA gyrase) and topoisomerase IV, which are required for bacterial DNA replication, transcription, repair, and recombination. Effects During Pregnancy and Lactation ◉ Summary of Use during Lactation No information is available on the clinical use of gatifloxacin during breastfeeding. Fluoroquinolones have traditionally not been used in infants because of concern about adverse effects on the infants' developing joints. However, recent studies indicate little risk. The calcium in milk might prevent absorption of the small amounts of fluoroquinolones in milk, but insufficient data exist to prove or disprove this assertion. Use of gatifloxacin is acceptable in nursing mothers with monitoring of the infant for possible effects on the gastrointestinal flora, such as diarrhea or candidiasis (thrush, diaper rash). However, it is preferable to use an alternate drug for which safety information is available. Maternal use of an ear drop or eye drop that contains gatifloxacin presents negligible risk for the nursing infant. To substantially diminish the amount of drug that reaches the breastmilk after using eye drops, place pressure over the tear duct by the corner of the eye for 1 minute or more, then remove the excess solution with an absorbent tissue. ◉ Effects in Breastfed Infants Relevant published information was not found as of the revision date. ◉ Effects on Lactation and Breastmilk Relevant published information was not found as of the revision date. Protein Binding 20% |
| 参考文献 |
|
| 其他信息 |
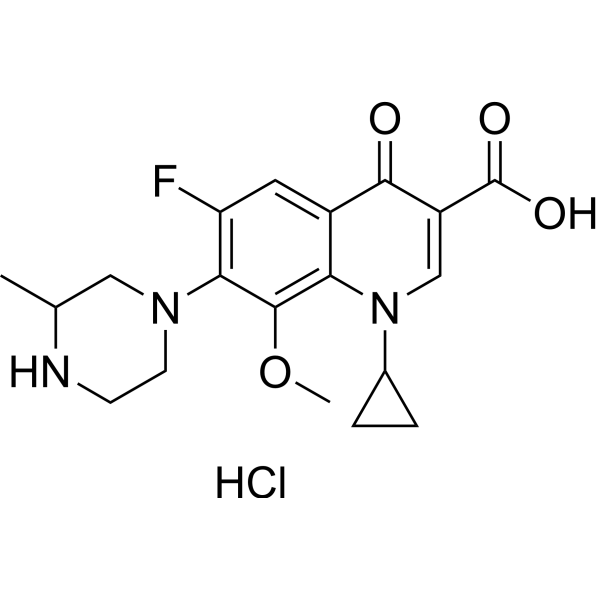
Gatifloxacin is a monocarboxylic acid that is 4-oxo-1,4-dihydroquinoline-3-carboxylic acid which is substituted on the nitrogen by a cyclopropyl group and at positions 6, 7, and 8 by fluoro, 3-methylpiperazin-1-yl, and methoxy groups, respectively. Gatifloxacin is an antibiotic of the fourth-generation fluoroquinolone family, that like other members of that family, inhibits the bacterial topoisomerase type-II enzymes. It has a role as an antiinfective agent, an EC 5.99.1.3 [DNA topoisomerase (ATP-hydrolysing)] inhibitor and an antimicrobial agent. It is a quinolinemonocarboxylic acid, a N-arylpiperazine, an organofluorine compound, a quinolone and a quinolone antibiotic.
Gatifloxacin is an antibiotic agent and a member of the fourth-generation fluoroquinolone family. It works by inhibiting the bacterial enzymes DNA gyrase and topoisomerase IV. It was first introduced by Bristol-Myers Squibb in 1999 under the brand name Tequin® for the treatment of respiratory tract infections. Gatifloxacin is available as tablets and in various aqueous solutions for intravenous therapy. It is also available as eye drops under the brand name Zymar® marketed by Allergan. The FDA withdrew its approval for the use of non-ophthalmic drug products containing gatifloxacin due to the high prevalence of gatifloxacin-associated dysglycemia adverse event reports and the high incidence of hyperglycemic and hypoglycemic episodes in patients taking gatifloxacin compared to those on macrolide antibiotics. Gatifloxacin anhydrous is a Quinolone Antimicrobial. Gatifloxacin is a synthetic 8-methoxyfluoroquinolone with antibacterial activity against a wide range of gram-negative and gram-positive microorganisms. Gatifloxacin exerts its effect through inhibition of DNA gyrase, an enzyme involved in DNA replication, transcription and repair, and inhibition of topoisomerase IV, an enzyme involved in partitioning of chromosomal DNA during bacterial cell division. Gatifloxacin is an antibiotic of the fourth-generation fluoroquinolone family, that like other members of that family, inhibits the bacterial enzymes DNA gyrase and topoisomerase IV. Bristol-Myers Squibb introduced Gatifloxacin in 1999 under the proprietary name Tequin™ for the treatment of respiratory tract infections, having licensed the medication from Kyorin Pharmaceutical Company of Japan. Allergan produces an eye-drop formulation called Zymar™. Gatifloxacin is available as tablets and in various aqueous solutions for intravenous therapy. [Wikipedia] A fluoroquinolone antibacterial agent and DNA TOPOISOMERASE II inhibitor that is used as an ophthalmic solution for the treatment of BACTERIAL CONJUNCTIVITIS. Drug Indication For the treatment of bronchitis, sinusitis, community-acquired pneumonia, and skin infections (abscesses, wounds) caused by S. pneumoniae, H. influenzae, S. aureus, M. pneumoniae, C. pneumoniae, L. pneumophila, S. pyogenes FDA Label Mechanism of Action The bactericidal action of Gatifloxacin results from inhibition of the enzymes topoisomerase II (DNA gyrase) and topoisomerase IV, which are required for bacterial DNA replication, transcription, repair, and recombination. Pharmacodynamics Gatifloxacin is a synthetic broad-spectrum 8-methoxyfluoroquinolone antibacterial agent for oral or intravenous administration. is bactericidal and its mode of action depends on blocking of bacterial DNA replication by binding itself to an enzyme called DNA gyrase, which allows the untwisting required to replicate one DNA double helix into two. Notably the drug has 100 times higher affinity for bacterial DNA gyrase than for mammalian. Gatifloxacin is a broad-spectrum antibiotic that is active against both Gram-positive and Gram-negative bacteria. It should be used only to treat or prevent infections that are proven or strongly suspected to be caused by bacteria. We determined the inhibitory activities of gatifloxacin against Staphylococcus aureus topoisomerase IV, Escherichia coli DNA gyrase, and HeLa cell topoisomerase II and compared them with those of several quinolones. The inhibitory activities of quinolones against these type II topoisomerases significantly correlated with their antibacterial activities or cytotoxicities (correlation coefficient [r] = 0.926 for S. aureus, r = 0.972 for E. coli, and r = 0.648 for HeLa cells). Gatifloxacin possessed potent inhibitory activities against bacterial type II topoisomerases (50% inhibitory concentration [IC50] = 13.8 microg/ml for S. aureus topoisomerase IV; IC50 = 0.109 microg/ml for E. coli DNA gyrase) but the lowest activity against HeLa cell topoisomerase II (IC50 = 265 microg/ml) among the quinolones tested. There was also a significant correlation between the inhibitory activities of quinolones against S. aureus topoisomerase IV and those against E. coli DNA gyrase (r = 0.969). However, the inhibitory activity against HeLa cell topoisomerase II did not correlate with that against either bacterial enzyme. The IC50 of gatifloxacin for HeLa cell topoisomerase II was 19 and was more than 2,400 times higher than that for S. aureus topoisomerase IV and that for E. coli DNA gyrase. These ratios were higher than those for other quinolones, indicating that gatifloxacin possesses a higher selectivity for bacterial type II topoisomerases. [1] Alternate mutations in the grlA and gyrA genes were observed through the first- to fourth-step mutants which were obtained from four Staphylococcus aureus strains by sequential selection with several fluoroquinolones. The increases in the MICs of gatifloxacin accompanying those mutational steps suggest that primary targets of gatifloxacin in the wild type and the first-, second-, and third-step mutants are wild-type topoisomerase IV (topo IV), wild-type DNA gyrase, singly mutated topo IV, and singly mutated DNA gyrase, respectively. Gatifloxacin had activity equal to that of tosufloxacin and activity more potent than those of norfloxacin, ofloxacin, ciprofloxacin, and sparfloxacin against the second-step mutants (grlA gyrA; gatifloxacin MIC range, 1.56 to 3.13 microg/ml) and had the most potent activity against the third-step mutants (grlA gyrA grlA; gatifloxacin MIC range, 1.56 to 6.25 microg/ml), suggesting that gatifloxacin possesses the most potent inhibitory activity against singly mutated topo IV and singly mutated DNA gyrase among the quinolones tested. Moreover, gatifloxacin selected resistant mutants from wild-type and the second-step mutants at a low frequency. Gatifloxacin possessed potent activity (MIC, 0.39 microg/ml) against the NorA-overproducing strain S. aureus NY12, the norA transformant, which was slightly lower than that against the parent strain SA113. The increases in the MICs of the quinolones tested against NY12 were negatively correlated with the hydrophobicity of the quinolones (correlation coefficient, -0.93; P < 0.01). Therefore, this slight decrease in the activity of gatifloxacin is attributable to its high hydrophobicity. Those properties of gatifloxacin likely explain its good activity against quinolone-resistant clinical isolates of S. aureus harboring the grlA, gyrA, and/or norA mutations. [2] Gatifloxacin can cause both hypoglycemia and hyperglycemia in both diabetic and non-diabetic patients. Gatifloxacin recently has been reported to stimulate insulin secretion by inhibition of ATP-sensitive K(+) (K(ATP)) channels in pancreatic beta-cells. Gatifloxacin-induced hypoglycemia is associated with concomitant use of sulfonylureas, and usually occurs immediately after administration of the drug. We find that gatifloxacin acutely stimulates insulin secretion from mouse pancreatic islets and that glibenclamide has additive effects on gatifloxacin-induced insulin secretion. On the other hand, gatifloxacin-induced hyperglycemia often takes several days to develop. We also demonstrate that chronic gatifloxacin treatment decreases islet insulin content by inhibiting insulin biosynthesis, which process may be associated with gatifloxacin-induced hyperglycemia. Moreover, discontinuation of gatifloxacin results in improved insulin secretory response. These data clarify the differing mechanisms of gatifloxacin-induced hyper- and hypoglycemia, and suggest that blood glucose levels should be carefully monitored during gatifloxacin administration, especially in elderly patients with renal insufficiency, unrecognized diabetes, or other metabolic disorders. Because the risk of potentially life-threatening dysglycemia is increased during gatifloxacin therapy, these findings have important implications for clinical practice. [3] In the present work, we evaluated the effect of gatifloxacin on the evolution of experimental murine infection with Nocardia brasiliensis using linezolid as a control. Gatifloxacin was injected subcutaneously at 100 mg/kg body weight every 8 h for 4 weeks. This compound was equally as efficient as linezolid in reducing the production of lesions. [4] |
| 分子式 |
C19H23CLFN3O4
|
|---|---|
| 分子量 |
411.8550
|
| 精确质量 |
411.136
|
| CAS号 |
121577-32-0
|
| 相关CAS号 |
Gatifloxacin;112811-59-3;Gatifloxacin-d3 hydrochloride; Gatifloxacin sesquihydrate; 121577-32-0; 316819-28-0 (mesylate); 180200-66-2 (sesquihydrate); 404858-36-2 (hemihydrate); 1190043-25-4; 121577-32-0; 1189946-71-1
|
| PubChem CID |
17956339
|
| 外观&性状 |
White to off-white solid powder
|
| tPSA |
82.1
|
| 氢键供体(HBD)数目 |
3
|
| 氢键受体(HBA)数目 |
8
|
| 可旋转键数目(RBC) |
4
|
| 重原子数目 |
28
|
| 分子复杂度/Complexity |
653
|
| 定义原子立体中心数目 |
0
|
| InChi Key |
GQYBNVXJQVIRGC-UHFFFAOYSA-N
|
| InChi Code |
InChI=1S/C19H22FN3O4.ClH/c1-10-8-22(6-5-21-10)16-14(20)7-12-15(18(16)27-2)23(11-3-4-11)9-13(17(12)24)19(25)26;/h7,9-11,21H,3-6,8H2,1-2H3,(H,25,26);1H
|
| 化学名 |
1-cyclopropyl-6-fluoro-8-methoxy-7-(3-methylpiperazin-1-yl)-4-oxoquinoline-3-carboxylic acid;hydrochloride
|
| 别名 |
gatifloxacin hydrochloride; 121577-32-0; Gatifloxacinacid; 160738-57-8; Gatifloxacin HCl; Gatifloxacin (hydrochloride); C3J4S9UD4E; Gatifloxacin hydrochloride [WHO-DD];
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month 注意: 请将本产品存放在密封且受保护的环境中,避免吸湿/受潮。 |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
H2O : ~10 mg/mL (~24.28 mM)
DMSO : ~4 mg/mL (~9.71 mM) |
|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: 6.67 mg/mL (16.19 mM) in PBS (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液; 超声助溶。 (<60°C).
请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.4280 mL | 12.1400 mL | 24.2801 mL | |
| 5 mM | 0.4856 mL | 2.4280 mL | 4.8560 mL | |
| 10 mM | 0.2428 mL | 1.2140 mL | 2.4280 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
| NCT Number | Recruitment | interventions | Conditions | Sponsor/Collaborators | Start Date | Phases |
| NCT00905762 | Completed | Drug: Gatifloxacin Drug: Moxifloxacin |
Healthy | Bausch & Lomb Incorporated | March 2009 | Phase 1 |
| NCT00410891 | Completed | Drug: gatifloxacin | Intravitreous Injections |
Stanford University | July 2008 | Phase 4 |
| NCT00464438 | Completed | Drug: moxifloxacin 0.5% eye drops Drug: gatifloxacin |
Bacterial Conjunctivitis | Allergan | June 2007 | Phase 4 |
| NCT00414011 | Completed | Drug: Moxifloxacin Drug: Gatifloxacin |
Epithelium, Corneal | Walter Reed Army Medical Center | February 2005 | Not Applicable |
| NCT00396084 | Completed | Drug: Gatifloxacin Drug: Isoniazid |
Tuberculosis | National Institute of Allergy and Infectious Diseases (NIAID) |
February 10, 2004 | Phase 1 Phase 2 |
 匹伐加宾
匹伐加宾
 磷酸氢化可的松
磷酸氢化可的松
 BAY-784
BAY-784
 Gly-PEG3-endo-BCN
Gly-PEG3-endo-BCN
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1





























