| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 10 mM * 1 mL in DMSO |
|
||
| 1mg |
|
||
| 5mg |
|
||
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| 500mg |
|
||
| Other Sizes |
|

| 靶点 |
Bcl-2 (Ki < 0.01 nM)
|
|---|---|
| 体外研究 (In Vitro) |
Gambogic Acid 是一种从 Garcinia hanburyi 中提取的笼状氧杂蒽酮,可抑制人类 Bcl-2 家族蛋白并激活半胱天冬酶,从而有效诱导多种癌细胞类型凋亡。此外,藤黄酸抑制 Kir2.1 通道,EC50 ≤ 100 nM。[1][2][3]在 nM 浓度下,藤黄酸可显着降低人脐静脉内皮细胞 (HUVEC) 的增殖、迁移、侵袭、管形成和微血管生长。 [4]
|
| 体内研究 (In Vivo) |
使用藤黄酸进行节律化疗,藤黄酸可有效抑制肿瘤血管生成并抑制肿瘤生长,且副作用低。 [4]藤黄酸具有多种有用的特性,例如诱导细胞凋亡、抑制增殖以及预防肿瘤血管生成和癌症转移。 [5]在动物肿瘤模型和人体临床试验中藤黄酸均能有效抑制肿瘤生长,且不良反应少,对免疫和造血系统毒性小。藤黄酸有可能引起肿瘤特异性毒性和组织特异性蛋白酶体抑制。 [6] 45 mg/kg (ip) 是小鼠的 LD50。 [7]
|
| 酶活实验 |
时间分辨荧光共振能量转移(TR-FRET)测定[1]
在TR-FRET实验中,将GST-Bcl-XL和抗gst -铽与FITC-Bad BH3肽在含0.005%的PBS中混合,每孔的总体积为20 μl,共96孔板。室温孵育30 min后,在含有10 nM Bcl-XL、10 nM FITC-Bad BH3肽和2 nM抗gst -terbium的反应混合物中加入2 μl含gambogic acid溶液,室温孵育30 min。用SpectraMax M5平板阅读器测量TR-FRET信号,使用以下设置:激发在330nm,发射FITC信号在490 nm,发射terbium信号在520 nm。 线粒体纯化和蛋白释放试验[1] 将HeLa细胞离心成球,然后在HM缓冲液(10 mM HEPES, pH 7.4, 250 mM甘露醇,10 mM KCl, 5 mM MgCl2, 1 mM EGTA)中洗涤一次,其中含有1 mM PMSF和蛋白酶抑制剂混合物。细胞颗粒然后在HM缓冲液中均匀,用b型杵进行50次均匀。匀浆600g离心2次,5min,去除细胞核和碎屑。将得到的上清液10000 g离心10分钟,用HM缓冲液洗涤2次,[1] 线粒体蛋白释放实验中,取10 μl线粒体(50 μl)加入到含有黄颡鱼酸的50 μl HM缓冲液中,将tBid或tBid与黄颡鱼酸或Bcl-2家族蛋白在30℃下预孵育15 min, 30℃下进一步孵育40-60 min,离心成粒,收集上清,在Laemmli样品缓冲液中煮沸,使用抗smac抗体进行SDS-PAGE/免疫印迹分析。 据报道,天然产物gambogic acid藤黄酸(GA)对培养的肿瘤细胞具有细胞毒活性,并在基于细胞的高通量筛选caspases(参与凋亡的蛋白酶)激活剂中被鉴定为一种活性化合物。利用抗凋亡Bcl-2家族蛋白Bfl-1作为筛选天然产物文库的靶标,我们在荧光偏振分析中发现GA是一种竞争性抑制剂,可以取代Bfl-1中的BH3肽。对BH3肽结合的竞争分析表明,GA在不同程度上抑制了所有6种人类Bcl-2家族蛋白,其中Mcl-1和Bcl-B被抑制的最有效[50%抑制所需浓度(IC(50)), < 1微mol/L]。利用时间分辨荧光共振能量转移实验也证实了BH3肽结合的竞争。通过离体线粒体实验表明,GA对抗凋亡的Bcl-2家族蛋白有抑制作用,重组纯化的Bcl-2家族蛋白在体外抑制SMAC的释放,表明GA以浓度依赖的方式中和了其对线粒体的抑制作用。GA通过凋亡机制杀死肿瘤细胞系,而在BH3肽位移处效力大大降低的GA类似物几乎没有细胞毒性活性。然而,在抗凋亡的Bcl-2家族蛋白缺乏细胞保护表型的bax-/-bak-/-细胞中,GA保留了细胞毒活性,这意味着GA还有其他靶点,有助于其细胞毒机制。总之,研究结果表明,抑制抗凋亡Bcl-2家族蛋白可能是GA杀死肿瘤细胞的细胞毒性机制之一。 藤黄酸(gambogic acid, GA)是一种从藤黄属(Garcinia hanburyi)树脂中提取的山酮,最近被证明可以结合转铁蛋白受体并表现出潜在的抗癌作用,其信号机制尚不完全清楚。由于NF-kappaB信号通路的关键作用,我们研究了GA对NF-kappaB介导的细胞反应和NF-kappaB调控的基因产物在人白血病癌细胞中的影响。GA增强肿瘤坏死因子(TNF)和化疗药物诱导的细胞凋亡,抑制抗凋亡基因产物(IAP1和IAP2、Bcl-2、Bcl-x(L)和TRAF1)、增殖(cyclin D1和c-Myc)、侵袭(COX-2和MMP-9)和血管生成(VEGF)的表达,所有这些基因产物都是由NF-kappaB调节的。GA抑制各种炎症剂和致癌物诱导的NF-kappaB活化,同时抑制TAK1/ tab1介导的IKK活化,抑制ikappabα磷酸化和降解,抑制p65磷酸化和核易位,最终消除NF-kappaB依赖性报告基因的表达。TNFR1、TRADD、TRAF2、NIK、TAK1/TAB1和IKKbeta诱导的NF-kappaB活化也被抑制。GA通过转铁蛋白受体介导的RNA干扰下调该受体的作用。 |
| 细胞实验 |
MTT 测定用于评估藤黄酸、CDDP 单独或两者一起等处理如何影响体外细胞活力。在 96 孔培养板中,接种细胞(2×104 个细胞/mL)。过夜孵育后,将藤黄酸按以下浓度应用于 NCI-H460、A549 和 NCI-H1299 细胞:0.125、0.25、0.25、0.5、1、2 和 4 μM、0.44、0.88、1.75、3.5、分别为 7、10.5 和 14 μM,以及 0.44、0.88、1.75、4、8、12 和 16 μM。在 NSCLC 细胞中,测试了三个序列的联合治疗:(a)藤黄酸,然后 CDDP 将细胞暴露于藤黄酸 48 小时,然后在洗去藤黄酸后,用 CDDP 处理细胞另外 48 小时; (b) CDDP,随后藤黄酸将细胞暴露于 CDDP 48 小时,然后在洗去 CDDP 后,用藤黄酸再处理细胞 48 小时; (c)同时处理的细胞暴露于藤黄酸和ADM 48小时。使用组合指数(CI)[2]分析药物相互作用的性质。
|
| 动物实验 |
Mice: A549 viable cells (5×106/100 μL PBS per mouse) are subcutaneously injected into the right flank of male SCID mice that are 7 to 8 weeks old in order to assess the in vivo antitumor activity of gambogic acid combined with CDDP. The mice are randomly assigned to one of four treatment groups when the tumor volume reaches 100 mm3, including control (saline only, n=5), gambogic acid (3.0 mg/kg every two days, intravenously; n=6), CDDP (4 mg/kg every week, intravenously; n=6), and sequential combination (CDDP treatment one day before gambogic acid treatment, n=6). To help detect any additive effects of combination therapy with platinum-based agents and gambogic acid, CDDP (4 mg/kg, weekly) is typically administered at doses lower than the maximum tolerated dose. Once every two days, a caliper is used to measure the tumor's size. Once every two days, body weight is measured. The tumors are removed after 14 days, and the mice are then put to death. They are then kept at -80°C for future research.
|
| 毒性/毒理 (Toxicokinetics/TK) |
rat LD50 intraperitoneal 88 mg/kg LIVER: OTHER CHANGES Indian Journal of Experimental Biology., 5(96), 1967 [PMID:6062434]
rat LD50 intravenous 107 mg/kg LIVER: OTHER CHANGES Indian Journal of Experimental Biology., 5(96), 1967 [PMID:6062434] mouse LD50 subcutaneous 354 mg/kg CRC Handbook of Antibiotic Compounds, Vols.1- , Berdy, J., Boca Raton, FL, CRC Press, 1980, 8(1)(331), 1982 mammal (species unspecified) LD50 unreported 55 mg/kg Zhongliu Yanjiu Cancer Review, Yu, R., et al., eds., Shanghai Science/Technology Publisher,Peop. Rep. China, 1994, -(220), 1994 |
| 参考文献 | |
| 其他信息 |
beta-Guttiferin has been reported in Garcinia hanburyi with data available.
Gambogic acid (2), a natural product isolated from the resin of Garcinia hurburyi tree, was discovered to be a potent apoptosis inducer using our cell- and caspase-based high-throughput screening assays. Gambogic acid was found to have an EC(50) of 0.78 microM in the caspase activation assay in T47D breast cancer cells. The apoptosis-inducing activity of gambogic acid was further characterized by a nuclear fragmentation assay and flow cytometry analysis in human breast tumor cells T47D. Gambogic acid was found to induce apoptosis independent of cell cycle, which is different from paclitaxel that arrests cells in the G2/M phase. To understand the structure-activity relationship (SAR) of gambogic acid, derivatives of 2 with modifications to different function groups were prepared. SAR studies of gambogic acid, as measured by the caspase activation assay, showed that the 9,10 carbon-carbon double bond of the alpha,beta-unsaturated ketone is important for biological activity, while the 6-hydroxy and 30-carboxy group can tolerate a variety of modifications. The importance of the 9,10 carbon-carbon double bond was confirmed by the traditional growth inhibition assay. The high potency of 2 as an inducer of apoptosis, its novel mechanism of action, easy isolation and abundant supply, as well as the fact that it is amenable to chemical modification, makes gambogic acid an attractive molecule for the development of anticancer agents.[2] Gambogic acid (GA), the main active compound of Gamboge hanburyi, has been previously reported to activate apoptosis in many types of cancer cell lines by targeting transferrin receptor and modulating nuclear factor-kappaB signaling pathway. Whether GA inhibits angiogenesis, which is crucial for cancer and other human diseases, remains unknown. Here, we found that GA significantly inhibited human umbilical vascular endothelial cell (HUVEC) proliferation, migration, invasion, tube formation, and microvessel growth at nanomolar concentration. In a xenograft prostate tumor model, we found that GA effectively inhibited tumor angiogenesis and suppressed tumor growth with low side effects using metronomic chemotherapy with GA. GA was more effective in activating apoptosis and inhibiting proliferation and migration in HUVECs than in human prostate cancer cells (PC3), suggesting GA might be a potential drug candidate in cancer therapy through angioprevention with low chemotoxicity. Furthermore, we showed that GA inhibited the activations of vascular endothelial growth factor receptor 2 and its downstream protein kinases, such as c-Src, focal adhesion kinase, and AKT. Together, these data suggest that GA inhibits angiogenesis and may be a viable drug candidate in antiangiogenesis and anticancer therapies. [4] Gambogic acid (GA) is a caged xanthone that is derived from Garcinia hanburyi and functions as a strong apoptotic inducer in many types of cancer cells. The distinct effectiveness of GA has led to its characterization as a novel anti-cancer agent. There is an increasing number of research studies focused on elucidating the molecular mechanisms of GA-induced anti-cancer effects, and several critical signaling pathways have been reported to be influenced by GA treatment. In this review, we summarize the multiple functional effects of GA administration in cancer cells including the induction of apoptosis, the inhibition of proliferation and the prevention of cancer metastasis and tumor angiogenesis. [5] |
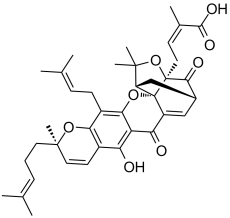
| 分子式 |
C38H44O8
|
|
|---|---|---|
| 分子量 |
628.75
|
|
| 精确质量 |
628.303
|
|
| 元素分析 |
C, 72.59; H, 7.05; O, 20.36
|
|
| CAS号 |
2752-65-0
|
|
| 相关CAS号 |
|
|
| PubChem CID |
9852185
|
|
| 外观&性状 |
Yellow solid powder
|
|
| 密度 |
1.3±0.1 g/cm3
|
|
| 沸点 |
808.9±65.0 °C at 760 mmHg
|
|
| 熔点 |
88.5°C
|
|
| 闪点 |
251.4±27.8 °C
|
|
| 蒸汽压 |
0.0±3.0 mmHg at 25°C
|
|
| 折射率 |
1.627
|
|
| LogP |
10.3
|
|
| tPSA |
119.36
|
|
| 氢键供体(HBD)数目 |
2
|
|
| 氢键受体(HBA)数目 |
8
|
|
| 可旋转键数目(RBC) |
8
|
|
| 重原子数目 |
46
|
|
| 分子复杂度/Complexity |
1490
|
|
| 定义原子立体中心数目 |
5
|
|
| SMILES |
O1C(C([H])([H])[H])(C([H])([H])[H])C2([H])C([H])([H])C3([H])C([H])=C4C(C5C(=C6C([H])=C([H])C(C([H])([H])[H])(C([H])([H])C([H])([H])/C(/[H])=C(\C([H])([H])[H])/C([H])([H])[H])OC6=C(C([H])([H])/C(/[H])=C(\C([H])([H])[H])/C([H])([H])[H])C=5OC24C1(C([H])([H])C([H])=C(C(=O)O[H])C([H])([H])[H])C3=O)O[H])=O
|
|
| InChi Key |
GEZHEQNLKAOMCA-RRZNCOCZSA-N
|
|
| InChi Code |
InChI=1S/C38H44O8/c1-20(2)10-9-15-36(8)16-14-24-29(39)28-30(40)26-18-23-19-27-35(6,7)46-37(33(23)41,17-13-22(5)34(42)43)38(26,27)45-32(28)25(31(24)44-36)12-11-21(3)4/h10-11,13-14,16,18,23,27,39H,9,12,15,17,19H2,1-8H3,(H,42,43)/b22-13-/t23-,27+,36-,37+,38-/m1/s1
|
|
| 化学名 |
(Z)-4-[(1S,2S,8R,17S,19R)-12-hydroxy-8,21,21-trimethyl-5-(3-methylbut-2-enyl)-8-(4-methylpent-3-enyl)-14,18-dioxo-3,7,20-trioxahexacyclo[15.4.1.02,15.02,19.04,13.06,11]docosa-4(13),5,9,11,15-pentaen-19-yl]-2-methylbut-2-enoic acid
|
|
| 别名 |
|
|
| HS Tariff Code |
2934.99.9001
|
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
|
|||
|---|---|---|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 2.5 mg/mL (3.98 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 25.0 mg/mL澄清DMSO储备液加入到400 μL PEG300中,混匀;然后向上述溶液中加入50 μL Tween-80,混匀;加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: ≥ 2.5 mg/mL (3.98 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 25.0 mg/mL 澄清 DMSO 储备液添加到 900 μL 玉米油中并混合均匀。 View More
配方 3 中的溶解度: 2% DMSO+40% PEG 300+2% Tween 80+ddH2O: 4mg/mL 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.5905 mL | 7.9523 mL | 15.9046 mL | |
| 5 mM | 0.3181 mL | 1.5905 mL | 3.1809 mL | |
| 10 mM | 0.1590 mL | 0.7952 mL | 1.5905 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
|
|---|
|
 |
 |
|---|
 |
 |
 IRF1-IN-1
IRF1-IN-1
 IRF1-IN-2
IRF1-IN-2
 Bfl-1-IN-6
Bfl-1-IN-6
 HDAC-IN-82
HDAC-IN-82
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1





























 COA
COA


