| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 5mg |
|
||
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| Other Sizes |
|
| 靶点 |
HIV
|
|---|---|
| 体外研究 (In Vitro) |
Fostemsavir/BMS-663068是小分子抑制剂BMS-626529的前药,通过与gp120结合并干扰病毒对CD4+ t细胞的附着来抑制人类免疫缺陷病毒1型(HIV-1)感染。由于gp120内的异质性,BMS-626529的活性依赖于病毒。为了更好地了解BMS-626529对HIV-1的抗谱,测定了其对多种实验室菌株和临床分离株的体外活性。BMS-626529对绝大多数病毒分离株的半数最大有效浓度(EC(50))值<10 nM;然而,对大多数易感病毒的敏感性变化大于6 log(10),在低pM范围内,对大多数易感病毒的有效浓度值达到最大值的一半。BMS-626529的体外抗病毒活性一般与嗜性或亚型无关,少数例外。测定BMS-626529对纯化gp120的结合亲和力表明,其抑制效力的一个因素可能是相对较长的解离半衰期。最后,在双药联合研究中,BMS-626529显示出与不同机制类别的抗逆转录病毒药物的加性或协同相互作用。这些结果表明BMS-626529应该对大多数HIV-1病毒有活性,并支持该化合物的持续临床开发。[1]
|
| 体内研究 (In Vivo) |
Fostemsavir Tris 对感染患者的药物敏感病毒表现出良好的抗病毒作用(IC50,<100 nM)[1]。血浆HIV-1 RNA载量较基线的最大中位数下降范围为1.21至1.73 log(10)拷贝/mL。BMS-626529的血浆浓度与抗病毒反应无关,而低基线抑制浓度和最低和平均稳态BMS-626529血浆浓度,当通过基线蛋白结合调整的90%抑制浓度(抑制商)调整时,与抗病毒反应相关。BMS-663068总体耐受良好。结论:联合或不联合利托那韦给药BMS-663068 8天可显著降低血浆HIV-1 RNA水平,并且通常耐受性良好。BMS-663068作为联合抗逆转录病毒治疗的一部分进行长期临床试验是有必要的。临床试验注册,nct01009814。 [J Infect Dis . 2012 Oct 1;206(7):1002-11]
|
| 酶活实验 |
Affinity and off-rates of attachment inhibitors from gp120.[1]
采用Micro BioSpin 6柱测定[3H]BMS-488043或[3H]BMS-626529与gp120的结合。结合溶液(30 μl)含有25 mM Tris-HCl (pH 7.5)、125 mM NaCl、50 nM gp120JRFL和连续稀释的[3H]BMS-488043或[3H]BMS-626529,使其达到平衡,然后吸附在MicroBioSpin 6柱上。将色谱柱离心(~ 14000 rpm) 5分钟,收集洗脱液,用闪烁计数器测定放射性。为了测量解离动力学,将150 nM [3H]BMS-626529或90 nM [3H]BMS-488043与60 nM gp120在室温下孵育1小时以达到平衡结合,然后加入大摩尔过量(14倍)可溶性CD4蛋白以驱动解离。在指定的时间间隔取等分,吸附到自旋柱上,离心,定量洗脱液中的放射性。通过比较有和没有可溶性CD4挑战的平行样品的氚信号,可以确定化合物结合的百分比。[1]
|
| 细胞实验 |
细胞毒性检测。[1]
在连续稀释的BMS-626529存在下进行细胞毒性实验长达6天,并使用XTT(2,3-双[2-甲氧基-4-硝基-5-巯基]- 2h -四氮唑-5-羧基苯胺)测定细胞活力。为了确定CC50值(杀死50%细胞所需的药物浓度),实验室适应细胞最初以0.1 × 106个细胞/ml的密度镀。在不含化合物的情况下,6天后细胞密度通常达到1.0 × 106 ~ 1.2 × 106/ml。 使用实验室病毒株进行药物敏感性试验。[1] MT-2(用于CXCR4或双向病毒)或PM1(用于ccr5向病毒)细胞以0.005的感染倍数感染,并在37°C的连续稀释药物中孵育4至6天。通过测定CXCR4病毒的逆转录酶(RT)活性或CCR5病毒的p24酶联免疫吸附试验来定量病毒产量。 用临床分离物进行药敏试验。[1] 临床分离菌感染pbmc微球,感染倍数为0.005,在0.5 ml体积中,37℃孵育3小时,然后在培养基中重悬,并添加到含有一系列稀释药物的板中。最终细胞密度为1 × 106个/ml。培养皿在37℃孵育,从感染后第5天开始使用p24酶联免疫吸附试验(ELISA)试剂盒监测病毒产量。当控制感染的上清液中p24水平在动态范围内(0.6 < A490 < 2.0)时,孵育终止。 用临床分离的包膜进行药敏试验。[1] 在百时美施贵宝赞助的试验中获得的血浆样本由Monogram生物科学公司进行测试,并与Monogram收集的其他样本一起进行测试。采用PhenoSense Entry法测定包膜的药物敏感性。通过逆转录酶PCR (RT-PCR)扩增包膜序列(gp160)并连接到pCXAS表达载体上。包膜表达载体制备成大的序列池(>200),以确保准确表示每个样本中存在的病毒准种的多样性。用包膜表达载体和在缺失包膜区域含有荧光素酶的复制缺陷HIV-1基因组载体共转染HEK293细胞,制备了重组HIV-1假病毒储备。利用重组假病毒颗粒感染表达CD4/CCR5/CXCR4的U87细胞系。通过比较BMS-626529存在和不存在时荧光素酶活性测定药物敏感性。药敏数据由Monogram Biosciences提供一半最大抑制浓度(IC50)值,并以此作为报告。 |
| 动物实验 |
Fifty HIV-1-infected subjects were randomized to 1 of 5 regimen groups (600 mg BMS-663068 plus 100 mg ritonavir every 12 hours [Q12H], 1200 mg BMS-663068 plus 100 mg ritonavir every bedtime, 1200 mg BMS-663068 plus 100 mg ritonavir Q12H, 1200 mg BMS-663068 Q12H plus 100 mg ritonavir every morning, or 1200 mg BMS-663068 Q12H) for 8 days in this open-label, multiple-dose, parallel study. The study assessed the pharmacodynamics, pharmacokinetics, and safety of BMS-663068.[J Infect Dis. 2012 Oct 1;206(7):1002-11.]
|
| 药代性质 (ADME/PK) |
Absorption
The absorption of temsavir is significantly limited by suboptimal dissolution and solubility following oral administration. Fostemsavir, a phosphonooxymethyl prodrug of temsavir, has improved aqueous solubility and stability under acidic conditions as compared to its parent drug - following oral administration of fostemsavir, the absolute bioavailability is approximately 26.9%. The Cmax and AUCtau following oral administration of fostemsavir 600mg twice daily was 1770 ng/mL and 12,900 ng.h/L, respectively, with a Tmax of approximately 2 hours. Co-administration of fostemsavir with a standard meal increases its AUC by approximately 10%, while co-administration with a high-fat meal increases its AUC by approximately 81%. Route of Elimination Temsavir is highly metabolized, after which it is excreted in the urine and feces as inactive metabolites. Approximately 51% of a given dose is excreted in the urine, with <2% comprising unchanged parent drug, and 33% is excreted in the feces, of which 1.1% is unchanged parent drug. Volume of Distribution The steady-state volume of distribution of temsavir following intravenous administration is approximately 29.5 L. Clearance The mean clearance and apparent clearance of temsavir, the active metabolite of fostemsavir, are 17.9 L/h and 66.4 L/h, respectively. Metabolism / Metabolites Fostemsavir is rapidly hydrolyzed to temsavir, its active metabolite, by alkaline phosphatase(s) present at the brush border membrane of the intestinal lumen. Temsavir undergoes further biotransformation to two predominant inactive metabolites: BMS-646915, a product of hydrolysis by esterases, and BMS-930644, an N-dealkylated metabolite generated via oxidation by CYP3A4. Approximately 36.1% of an administered oral dose is metabolized by esterases, 21.2% is metabolized by CYP3A4, and <1% is conjugated by UDP-glucuronosyltransferases (UGT) prior to elimination. Both temsavir and its two predominant metabolites are known to inhibit BCRP. Biological Half-Life The half-life of temsavir is approximately 11 hours. Fostemsavir is generally undetectable in plasma following oral administration. |
| 毒性/毒理 (Toxicokinetics/TK) |
Effects During Pregnancy and Lactation
◉ Summary of Use during Lactation No information is available on the use of fostemsavir during breastfeeding. Because the drug and its active metabolite temsavir are over 80% protein bound, the amounts in milk are likely to be low. Achieving and maintaining viral suppression with antiretroviral therapy decreases breastfeeding transmission risk to less than 1%, but not zero. Individuals with HIV who are on antiretroviral therapy with a sustained undetectable viral load and who choose to breastfeed should be supported in this decision. If a viral load is not suppressed, banked pasteurized donor milk or formula is recommended. ◉ Effects in Breastfed Infants Relevant published information was not found as of the revision date. ◉ Effects on Lactation and Breastmilk Relevant published information was not found as of the revision date. |
| 参考文献 | |
| 其他信息 |
Fostemsavir (brand name: Rukobia) is a prescription medicine approved by the U.S. Food and Drug Administration (FDA) for the treatment of HIV infection in treatment-experienced adults who meet certain requirements, as determined by a health care provider. Fostemsavir is always used in combination with other HIV medicines.
Fostemsavir Tromethamine is the tromethamine salt form of fostemsavir, an orally bioavailable phosphonooxymethyl prodrug of the human immunodeficiency virus type 1 (HIV-1) attachment inhibitor temsavir, with activity against HIV. Upon oral administration, fostemsavir is hydrolyzed to the active moiety temsavir. Temsavir targets and binds to the gp120 subunit within the HIV-1 envelope glycoprotein gp160. This selectively inhibits the interaction between HIV-1 virus and host cellular CD4 receptors, thereby preventing HIV-1 virus attachment. This also inhibits gp120-dependent post-attachment steps that are needed for HIV-1 viral entry into host cells. Drug Indication Rukobia, in combination with other antiretrovirals, is indicated for the treatment of adults with multidrug resistant HIV-1 infection for whom it is otherwise not possible to construct a suppressive anti-viral regimen. |
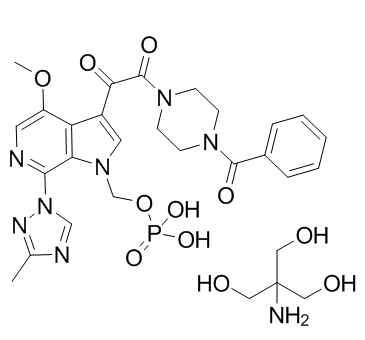
| 分子式 |
C29H37N8O11P
|
|---|---|
| 分子量 |
704.6248
|
| 精确质量 |
704.231
|
| 元素分析 |
C, 49.43; H, 5.29; N, 15.90; O, 24.98; P, 4.40
|
| CAS号 |
864953-39-9
|
| 相关CAS号 |
Temsavir;701213-36-7; 864953-29-7(free base); 864953-39-9 (tromethamine) ; 864953-31-1 (disodium); 942117-71-7 (dihydrate)
|
| PubChem CID |
46892186
|
| 外观&性状 |
White to off-white solid powder
|
| tPSA |
269Ų
|
| 氢键供体(HBD)数目 |
6
|
| 氢键受体(HBA)数目 |
15
|
| 可旋转键数目(RBC) |
11
|
| 重原子数目 |
49
|
| 分子复杂度/Complexity |
1070
|
| 定义原子立体中心数目 |
0
|
| SMILES |
O=C(C1C2C(=C(N3C=NC(C)=N3)N=CC=2OC)N(COP(O)(O)=O)C=1)C(N1CCN(C(C2C=CC=CC=2)=O)CC1)=O.OCC(CO)(CO)N
|
| InChi Key |
RRGJSMBMTOKHTE-UHFFFAOYSA-N
|
| InChi Code |
InChI=1S/C25H26N7O8P.C4H11NO3/c1-16-27-14-32(28-16)23-21-20(19(39-2)12-26-23)18(13-31(21)15-40-41(36,37)38)22(33)25(35)30-10-8-29(9-11-30)24(34)17-6-4-3-5-7-17;5-4(1-6,2-7)3-8/h3-7,12-14H,8-11,15H2,1-2H3,(H2,36,37,38);6-8H,1-3,5H2
|
| 化学名 |
1,2-Ethanedione, 1-(4-benzoyl-1-piperazinyl)-2-(4-methoxy-7-(3-methyl-1H-1,2,4-triazol-1-yl)-1-((phosphonooxy)methyl)-1H-pyrrolo(2,3-c)pyridin-3-yl)-, compd. with 2-amino-2-(hydroxymethyl)-1,3-propanediol (1:1)
|
| 别名 |
Fostemsavir tromethamine; BMS663068; 864953-39-9; Fostemsavir tromethamine; Fostemsavir Tris; BMS-663068 Tris; BMS 663068 (Tris); BMS-663068 (Tris); Fostemsavir trometamol; Rukobia; BMS-663068; BMS 663068; BMS-663068-03; GSK3684934A
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month 注意: 请将本产品存放在密封且受保护的环境中,避免吸湿/受潮。 |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
DMSO : ~125 mg/mL (~177.40 mM)
H2O : ~100 mg/mL (~141.92 mM) |
|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 2.08 mg/mL (2.95 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 20.8 mg/mL澄清DMSO储备液加入400 μL PEG300中,混匀;然后向上述溶液中加入50 μL Tween-80,混匀;加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: ≥ 2.08 mg/mL (2.95 mM) (饱和度未知) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 20.8 mg/mL澄清DMSO储备液加入900 μL 20% SBE-β-CD生理盐水溶液中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 View More
配方 3 中的溶解度: ≥ 2.08 mg/mL (2.95 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 配方 4 中的溶解度: 50 mg/mL (70.96 mM) in PBS (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液; 超声助溶. 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.4192 mL | 7.0960 mL | 14.1920 mL | |
| 5 mM | 0.2838 mL | 1.4192 mL | 2.8384 mL | |
| 10 mM | 0.1419 mL | 0.7096 mL | 1.4192 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
 HIV-1 inhibitor-80
HIV-1 inhibitor-80
 Antitumor agent-191
Antitumor agent-191
 HIV-1 inhibitor-79
HIV-1 inhibitor-79
 2,4-Dihydroxypyridine
2,4-Dihydroxypyridine
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1






























