| 规格 | 价格 | |
|---|---|---|
| 500mg | ||
| 1g | ||
| Other Sizes |
| 体外研究 (In Vitro) |
福辛普利(0-100 μM;30 分钟)部分抑制脂质体和重组 LPLA2 的共沉降 [1]。福辛普利 (250 nM) 对 LPLA2 可溶性酯酶活性没有抑制作用 [1]。 ACE 活性被福辛普利(0.372、0.744、1.116 μM)非竞争性抑制,Ki 值为 1.675 μM [2]。
|
|---|---|
| 体内研究 (In Vivo) |
给予福辛普利(4.67 mg/kg;口服;4 周)的大鼠可降低肌酸激酶 (CK) 和乳酸脱氢酶 (LDH) 的水平,并可防止结构改变和心脏功能障碍 [3]。在AMI大鼠模型中,福辛普利(4.67 mg/kg;口服;4周)可以抑制cleaved-caspase 3的产生和心肌细胞凋亡[3]。
|
| 动物实验 |
Animal/Disease Models: Acute myocardial infarction (AMI) rat model after heart failure (SPF grade SD (SD (Sprague-Dawley)) rat, 265±15g) [3]
Doses: 4.67mg/kg Route of Administration: po (oral gavage); 4 weeks Experimental Results: Inhibition of cardiac dysfunction and structural changes and inhibition of apoptosis. |
| 药代性质 (ADME/PK) |
Absorption, Distribution and Excretion
Average absolute absorption is 36%. The primary site of absorption is the proximal small intestine (duodenum/jejunum). Food slows the rate of absorption with no effect on the extent of absorption. After oral administration of radiolabeled fosinopril, approximately half of the absorbed dose is excreted in the urine and the remainder is excreted in the feces. 26 - 39 mL/min [healthy] Metabolism / Metabolites Since fosinoprilat is not biotransformed after intravenous administration, fosinopril, not fosinoprilat, appears to be the precursor for the glucuronide and p-hydroxy metabolites. Biological Half-Life 12 hours |
| 毒性/毒理 (Toxicokinetics/TK) |
Hepatotoxicity
Fosinopril, like other ACE inhibitors, has been associated with a low rate of serum aminotransferase elevations ( Likelihood score: D (possible rare cause of clinically apparent liver injury). Effects During Pregnancy and Lactation ◉ Summary of Use during Lactation Because no information is available on the use of fosinopril during breastfeeding, an alternate drug may be preferred, especially while nursing a newborn or preterm infant. ◉ Effects in Breastfed Infants Relevant published information was not found as of the revision date. ◉ Effects on Lactation and Breastmilk Relevant published information was not found as of the revision date. Protein Binding Fosinoprilat is ≥95% protein bound |
| 参考文献 |
|
| 其他信息 |
Pharmacodynamics
Following oral administration, fosinopril is rapidly and completely hydrolyzed to its principle active metabolite, fosinoprilat. Hydrolysis is thought to occur in the gastrointestinal mucosa and liver. Fosinoprilat is a competitive inhibitor of ACE, a peptidyl dipeptidase that is part of the RAAS. The RAAS is a homeostatic mechanism for regulating hemodynamics, water and electrolyte balance. During sympathetic stimulation or when renal blood pressure or blood flow is reduced, renin is released from the granular cells of the juxtaglomerular apparatus in the kidneys. In the blood stream, renin cleaves circulating angiotensinogen to ATI, which is subsequently cleaved to ATII by ACE. ATII increases blood pressure using a number of mechanisms. First, it stimulates the secretion of aldosterone from the adrenal cortex. Aldosterone travels to the distal convoluted tubule (DCT) and collecting tubule of nephrons where it increases sodium and water reabsorption by increasing the number of sodium channels and sodium-potassium ATPases on cell membranes. Second, ATII stimulates the secretion of vasopressin (also known as antidiuretic hormone or ADH) from the posterior pituitary gland. ADH stimulates further water reabsorption from the kidneys via insertion of aquaporin-2 channels on the apical surface of cells of the DCT and collecting tubules. Third, ATII increases blood pressure through direct arterial vasoconstriction. Stimulation of the Type 1 ATII receptor on vascular smooth muscle cells leads to a cascade of events resulting in myocyte contraction and vasoconstriction. In addition to these major effects, ATII induces the thirst response via stimulation of hypothalamic neurons. ACE inhibitors inhibit the rapid conversion of ATI to ATII and antagonize RAAS-induced increases in blood pressure. ACE (also known as kininase II) is also involved in the enzymatic deactivation of bradykinin, a vasodilator. Inhibiting the deactivation of bradykinin increases bradykinin levels and may further sustain the effects of fosinoprilat by causing increased vasodilation and decreased blood pressure. |
| 分子式 |
C30H46NO7P
|
|---|---|
| 分子量 |
563.66254
|
| 精确质量 |
563.301
|
| CAS号 |
98048-97-6
|
| 相关CAS号 |
Fosinopril sodium;88889-14-9
|
| PubChem CID |
55891
|
| 外观&性状 |
Typically exists as solid at room temperature
|
| 密度 |
1.173 g/cm3
|
| 沸点 |
705.7ºC at 760 mmHg
|
| 熔点 |
149-153ºC
|
| 闪点 |
380.6ºC
|
| 折射率 |
1.531
|
| LogP |
6.059
|
| tPSA |
120.02
|
| 氢键供体(HBD)数目 |
1
|
| 氢键受体(HBA)数目 |
7
|
| 可旋转键数目(RBC) |
15
|
| 重原子数目 |
39
|
| 分子复杂度/Complexity |
850
|
| 定义原子立体中心数目 |
2
|
| SMILES |
CCC(=O)OC(C(C)C)OP(=O)(CCCCC1=CC=CC=C1)CC(=O)N2C[C@@H](C[C@H]2C(=O)O)C3CCCCC3
|
| InChi Key |
BIDNLKIUORFRQP-KKDZQFHASA-N
|
| InChi Code |
InChI=1S/C30H46NO7P/c1-4-28(33)37-30(22(2)3)38-39(36,18-12-11-15-23-13-7-5-8-14-23)21-27(32)31-20-25(19-26(31)29(34)35)24-16-9-6-10-17-24/h5,7-8,13-14,22,24-26,30H,4,6,9-12,15-21H2,1-3H3,(H,34,35)/t25-,26+,30?,39?/m0/s1
|
| 化学名 |
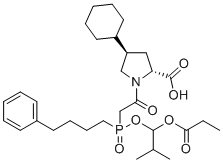
(2R,4R)-4-cyclohexyl-1-(2-((2-methyl-1-(propionyloxy)propoxy)(4-phenylbutyl)phosphoryl)acetyl)pyrrolidine-2-carboxylic
acid
|
| 别名 |
SQ-28555SQ28555SQ 28555
FosinoprilMonopril
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples
|
|---|---|
| 溶解度 (体内实验) |
注意: 如下所列的是一些常用的体内动物实验溶解配方,主要用于溶解难溶或不溶于水的产品(水溶度<1 mg/mL)。 建议您先取少量样品进行尝试,如该配方可行,再根据实验需求增加样品量。
注射用配方
注射用配方1: DMSO : Tween 80: Saline = 10 : 5 : 85 (如: 100 μL DMSO → 50 μL Tween 80 → 850 μL Saline)(IP/IV/IM/SC等) *生理盐水/Saline的制备:将0.9g氯化钠/NaCl溶解在100 mL ddH ₂ O中,得到澄清溶液。 注射用配方 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (如: 100 μL DMSO → 400 μL PEG300 → 50 μL Tween 80 → 450 μL Saline) 注射用配方 3: DMSO : Corn oil = 10 : 90 (如: 100 μL DMSO → 900 μL Corn oil) 示例: 以注射用配方 3 (DMSO : Corn oil = 10 : 90) 为例说明, 如果要配制 1 mL 2.5 mg/mL的工作液, 您可以取 100 μL 25 mg/mL 澄清的 DMSO 储备液,加到 900 μL Corn oil/玉米油中, 混合均匀。 View More
注射用配方 4: DMSO : 20% SBE-β-CD in Saline = 10 : 90 [如:100 μL DMSO → 900 μL (20% SBE-β-CD in Saline)] 口服配方
口服配方 1: 悬浮于0.5% CMC Na (羧甲基纤维素钠) 口服配方 2: 悬浮于0.5% Carboxymethyl cellulose (羧甲基纤维素) 示例: 以口服配方 1 (悬浮于 0.5% CMC Na)为例说明, 如果要配制 100 mL 2.5 mg/mL 的工作液, 您可以先取0.5g CMC Na并将其溶解于100mL ddH2O中,得到0.5%CMC-Na澄清溶液;然后将250 mg待测化合物加到100 mL前述 0.5%CMC Na溶液中,得到悬浮液。 View More
口服配方 3: 溶解于 PEG400 (聚乙二醇400) 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.7741 mL | 8.8706 mL | 17.7412 mL | |
| 5 mM | 0.3548 mL | 1.7741 mL | 3.5482 mL | |
| 10 mM | 0.1774 mL | 0.8871 mL | 1.7741 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1






























 COA
COA




