
| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 100mg |
|
||
| 250mg |
|
||
| 500mg |
|
||
| 1g |
|
||
| 5g |
|
||
| Other Sizes |
|

| 靶点 |
Xanthine oxidase (XO) (Ki = 0.6 nM)
|
||
|---|---|---|---|
| 体外研究 (In Vitro) |
Ki 和 Ki' 值分别为 0.6 nM 和 3.1 nM,显示出对氧化型和还原型黄嘌呤氧化酶的抑制作用,非布索坦对纯牛乳黄嘌呤氧化酶的活性表现出强烈的混合型抑制作用[1]。
嘌呤类似物别嘌呤醇作为黄嘌呤氧化酶(XO)抑制剂在治疗高尿酸血症和痛风方面的临床应用已有30多年。然而,由于与嘌呤化合物结构相似,别嘌呤醇及其主要活性产物氧嘌呤醇及其各自的代谢产物抑制了参与嘌呤和嘧啶代谢的其他酶。非布索坦(TEI-6720,TMX-67)是一种强效的XO非嘌呤抑制剂,目前正在进行临床评估,用于治疗高尿酸血症和痛风。在这项研究中,我们研究了非布索坦对嘌呤和嘧啶代谢中几种酶的影响,并表征了非布索坦抑制XO活性的机制。非布索坦对纯化牛乳XO的活性表现出强烈的混合型抑制作用,Ki和Ki'值分别为0.6和3.1 nM,表明对氧化和还原形式的XO都有抑制作用。相比之下,在高达100μM的浓度下,非布索坦对以下嘌呤和嘧啶代谢酶的活性没有显著影响:鸟嘌呤脱氨酶、次黄嘌呤鸟嘌呤磷酸核糖转移酶、嘌呤核苷磷酸化酶、乳清酸磷酸核糖基转移酶和乳清酸-5'-单磷酸脱羧酶。这些结果表明,非布索坦是一种有效的非嘌呤选择性XO抑制剂,可用于治疗高尿酸血症和痛风[1]。 |
||
| 体内研究 (In Vivo) |
与果糖+P大鼠相比,给予高果糖饮食(即60%果糖)8周的大鼠在给予非布索坦(5-6mg/kg;即,每天一次,持续 4 周)。然而,给予正常饮食的大鼠单独使用非布索坦治疗并没有表现出明显的效果[2]。在患有或不患有并发高尿酸血症的 5/6 Nx(5/6 肾切除术)大鼠中,非布索坦(3-4 mg/kg;口服;每天 4 周)联合氧酸(750 mg/kg;口服灌胃;每天 4 周)周)可预防肾损伤[3]。非布索坦(2.5 mg/kg;口服;每天服用,持续 12 周)可降低动脉粥样硬化动物主动脉壁中 ROS 的含量,并防止 ApoE−/− 小鼠中斑块的形成[4]。 fruxostat(15.6 mg/kg;口服;每天一次,持续 21 天)的抗抑郁作用通过小鼠强迫游泳试验 (FST) 中不动时间的大幅减少得到证实 [5]。当与多柔比星(10 mg/kg;口服;每日给药,持续 21 天)联合给药时,fruxostat(10 mg/kg;口服;口服;每日给药,持续 21 天)显着降低肾毒性指标和炎症介质,将氧化应激生物标志物水平恢复至正常,并抑制肾 caspase-3 的表达[6]。
果糖摄入增加与高尿酸血症、代谢综合征和肾损伤有关。本研究评估了非布索坦(Fx),一种研究性非嘌呤和选择性黄嘌呤氧化酶抑制剂,是否可以缓解代谢综合征的特征以及高果糖饮食引起的大鼠肾脏血流动力学改变和传入动脉病变。两组大鼠喂食高果糖饮食(60%果糖)8周,两组接受正常饮食。对于每种饮食,一组在最后4周(即代谢综合征发作后)服用Fx(5-6mg/kg(-1).天(-1)的饮用水),另一组不接受治疗(安慰剂;P)。每天测量体重。在基线、4周和8周时测量收缩压和空腹血浆尿酸(UA)、胰岛素和甘油三酯。研究结束时,对肾脏血流动力学和组织形态学进行了评估。高果糖饮食与高尿酸血症、高血压以及血浆甘油三酯和胰岛素升高有关。与果糖+P相比,果糖+Fx大鼠的血压、UA、甘油三酯和胰岛素显著降低(所有比较均P<0.05)。此外,与果糖+P大鼠相比,果糖+Fx大鼠的肾小球压力、肾血管收缩和传入小动脉面积显著降低。对正常饮食的大鼠进行Fx治疗没有显著影响。总之,代谢综合征大鼠血浆UA与Fx的正常化缓解了代谢和肾小球血流动力学和形态学改变。这些结果为高尿酸血症在果糖介导的代谢综合征中的致病作用提供了进一步的证据。[2] 结果:与5/6 Nx+V+P相比,5/6 Nx+OA+P大鼠出现了高尿酸血症、肾血管收缩和肾小球高血压,与传入动脉病变的进一步加重有关。Fx预防了5/6 Nx+OA+Fx大鼠的高尿酸血症,改善了5/6 Nx+V+Fx和5/6 Nx+OA+Fx组的蛋白尿,保护了肾功能,预防了肾小球高血压。功能改善伴随着传入小动脉形态的保留和肾小管间质纤维化的减少。 结论:Fx可预防5/6 Nx大鼠合并和不合并高尿酸血症的肾损伤。由于Fx有助于保持肾小球前血管形态,即使在存在全身性高血压的情况下,正常的肾小球压力也得以维持。[3] 动脉粥样硬化是一种由动脉壁脂质沉积引起的慢性炎症性疾病。多种机制参与炎症过程,包括氧化应激。黄嘌呤氧化酶(XO)是活性氧(ROS)的主要来源,与动脉粥样硬化的发病机制有关,但其潜在机制尚不清楚。在这里,我们发现ApoE(-/-)小鼠动脉粥样硬化斑块中巨噬细胞和主动脉内皮细胞中的XO表达增强,并且强效XO抑制剂非布索坦抑制了ApoE(//-)小鼠的斑块形成,降低了动脉ROS水平,改善了内皮功能障碍,而不影响血浆胆固醇水平。在体外,非布索坦抑制了胆固醇晶体诱导的小鼠巨噬细胞中ROS的形成和炎性细胞因子的释放。这些结果表明,在动脉粥样硬化斑块中,XO介导的ROS形成是促炎的,非布司他对XO的抑制是动脉粥样硬化的一种潜在治疗方法。[4] 别嘌呤醇和非布索坦表现出显著的抗抑郁样作用,与对照组相比,FST小鼠的不动期缩短。别嘌呤醇和非布索坦的疗效与氟西汀相当。 结论:别嘌呤醇和非布索坦具有明显的抗抑郁作用。[5] 西地那非和非布索坦可预防阿霉素诱导的肾毒性;然而,确切的机制仍有待阐明。研究了西地那非和非布索坦对阿霉素诱导的大鼠肾毒性的影响。雄性大鼠被分为九组。第一组作为正常对照组,第二组接受50%二甲亚砜(DMSO)治疗,第三组接受阿霉素(3.5mg/kg,i.p.)治疗,每周两次,持续3周。接下来的3组分别每天服用西地那非(5mg/kg;口服)、非布索坦(10mg/kg;口服)及其组合,持续21天。最后3组接受阿霉素与西地那非、非布索坦或其组合的联合治疗。通过光学显微镜进行组织病理学评估,并通过测量以下参数进行生化评估:肾功能生物标志物[血清尿素、肌酐和尿酸水平],氧化应激生物标志物[GSH和MDA的肾脏含量],凋亡标志物即;肾组织中的半胱氨酸天冬氨酸蛋白酶-3和炎症介质肿瘤坏死因子α(TNF-α)。阿霉素诱导肾毒性标志物、半胱氨酸天冬氨酸蛋白酶-3表达显著升高,并诱导炎症和氧化应激。肾脏的组织学变化为肾小管坏死。西地那非和/或非布索坦与阿霉素联合给药后,肾毒性标志物和炎症介质显著降低,氧化应激生物标志物恢复正常值,并阻碍肾脏半胱氨酸天冬氨酸蛋白酶-3的表达。它们还改善了阿霉素诱导的组织学变化。西地那非和非布索坦通过改善阿霉素诱导的生化、炎症、组织病理学和免疫组织化学改变,是对抗阿霉素肾毒性的有前景的保护剂[6]。 |
||
| 酶活实验 |
酶活性测定[1]
所有酶活性测定均使用日立分光光度计(型号U-3200)进行,该分光光度仪带有6细胞定位器,细胞温度保持在25°C。对于所有测定,在具有1.0-cm光路的3-mL池中,反应混合物的最终体积为2.5mL。 XO检测[1] 该测定如前所述进行(Osada等人,1993)。反应混合物含有0.1 M磷酸钠缓冲液(pH 7.4)、黄嘌呤(2.5-20μM)和XO(1.1μg蛋白质)。通过加入酶开始反应,在292 nm处形成尿酸(黄嘌呤→尿酸)。酶活性计算为反应初始线性部分每分钟每毫克蛋白质形成的μmol尿酸。计算中使用的尿酸的摩尔消光系数(Δɛ292)为10923 M−1cm−1。当研究非布索坦对XO活性的抑制作用时,黄嘌呤的浓度在2.5至20μM之间变化,非布索他汀的浓度在0至1.5 nM之间。抑制机制由Lineweaver-Burk图确定,Ki和Ki'值分别由Dixon图和1/V轴截距重样计算。 鸟嘌呤脱氨酶测定[1] 该测定程序基于Lewis和Glantz(1974)的工作。反应混合物含有0.2 M磷酸钠缓冲液(pH 7.0)、15μM鸟嘌呤[底物浓度接近12.5μM的米氏常数(Glantz和Lewis,1978)]和鸟嘌呤脱氨酶(0.4μg蛋白质)。充分混合后,在246 nm处监测鸟嘌呤(鸟嘌呤→黄嘌呤)的消耗。在反应的初始线性部分,酶活性以每分钟每毫克蛋白质形成的nmol黄嘌呤计算。计算中使用的黄嘌呤的摩尔消光系数(Δɛ246)为5662 M−1cm−1。 HGPRT测定[1] HGPRT活性的测定方法是对Giacomello和Salerno(1978)方法的改进。反应混合物含有50 mM Tris-HCl缓冲液(pH 8.0)、2 mM MgCl2、0.5 mM PRPP、1 mM DTT、10μM次黄嘌呤[接近7.7μM米氏常数的底物浓度(Giacomello和Salerno,1978)]和HGPRT(7.1μg蛋白质)。充分混合后,监测肌苷-5′-单磷酸(IMP)(次黄嘌呤→IMP)形成导致的245 nm处吸光度的增加。在反应的初始线性部分,使用IMP的摩尔消光系数(Δɛ245)1657 M−1cm−1,以每分钟每毫克蛋白质形成的nmol IMP计算酶活性。 PNP测定[1] 该测定采用Stoeckler和Parks(1985)描述的方法进行。反应混合物含有0.5 M磷酸钾缓冲液(pH 7.5)、50μM鸟苷[接近32μM米氏常数的底物浓度(Stoeckler和Parks,1985)]、1 mM DTT和PNP(0.8 mg蛋白质)。充分混合后,监测了由于鸟苷的消耗而导致的258 nm处吸光度的降低(鸟苷→鸟嘌呤)。酶活性计算为反应初始线性部分每分钟每毫克蛋白质形成的μmol鸟嘌呤。本次计算中使用的鸟嘌呤的摩尔消光系数(Δɛ258)为5911 M−1cm−1。 OPRT检测[1] 利用Lieberman等人(1955)的方法的改进来测定OPRT活性。反应混合物含有50 mM Tris-HCl(pH 8.0)、2 mM MgCl2、1 mM DTT、15μM OA[接近15.4μM米氏常数的底物浓度(Shostak等人,1990)]、0.5 mM PRPP和OPRT(4.7μg蛋白质)。充分混合后,监测295 nm处光密度的降低,反映OA的消耗(OA→OMP)。在反应的初始线性部分,酶活性以每分钟每毫克蛋白质形成的OMP nmol计算。OMP(2997 M−1 cm−1)的摩尔消光系数(Δɛ295)用于计算酶活性。 OMPDC测定[1] OMPDC活性测定采用Lieberman等人(1955)方法的改进进行。反应混合物含有50 mM Tris-HCl(pH 8.0)、1 mM DTT、10μM OMP[接近6μM米氏常数的底物浓度(Shostak等人,1990)]和OMPDC(10μg蛋白质)。充分混合后,监测285nm处吸光度的降低,反映了OMP的消耗[OMP→尿苷-5′-单磷酸盐(UMP)]。在反应的初始线性部分,酶活性以每分钟每毫克蛋白质形成的nmol UMP计算。UMP(2285M-1cm-1)的摩尔消光系数Δɛ285用于计算酶活性。 |
||
| 动物实验 |
|
||
| 药代性质 (ADME/PK) |
Absorption
After oral administration, about 85% of febuxostat is absorbed rapidly. Tmax ranges from 1 to 1.5 hours. Following once-daily oral administration, Cmax was approximately 1.6 ± 0.6 mcg/mL at a dose of 40 mg febuxostat and 2.6 ± 1.7 mcg/mL at a dose of 80 mg febuxostat. A high-fat meal decreased Cmax by 49% and AUC by 18%, but there were no clinically significant changes in the ability of febuxostat to decrease serum uric acid concentrations. Route of Elimination Febuxostat is eliminated via both hepatic and renal pathways. Following oral administration of 80 mg radiolabeled febuxostat, approximately 49% of the dose was recovered in the urine. In urine, about 3% of the recovered dose accounted for unchanged febuxostat, 30% accounted for the acyl glucuronide metabolite, 13% accounted for oxidative metabolites and their conjugates, and 3% accounted for unidentified metabolites. Approximately 45% of the total dose was recovered in the feces, where 12% of the dose accounted for the unchanged parent drug. About 1% accounted for the acyl glucuronide metabolite, 25% accounted for oxidative metabolites and their conjugates, and 7% accounted for unidentified metabolites. Volume of Distribution The apparent steady-state volume of distribution (Vss/F) of febuxostat ranges from 29 to 75 L, indicating a low to medium volume of distribution. Clearance Following oral administration of single doses of 10 to 240 mg, the mean apparent total clearance ranged from 10 to 12 L/h. Metabolism / Metabolites Febuxostat is metabolized in the liver by UDP-glucuronosyltransferase (UGT) and Cytochrome P450 (CYP) enzymes, with the relative contribution of each enzyme isoform in the metabolism of febuxostat not fully elucidated. UGT1A1, UGT1A3, UGT1A9, and UGT2B7 mediate conjugation of febuxostat, which approximately accounts for 22–44% of the metabolism of the total dose administered, to produce the acyl-glucuronide metabolite. CYP1A2, CYP2C8, CYP2C9, and non-P450 enzymes are responsible for the oxidation reaction, which accounts for 2-8% of the metabolism of the dose. Oxidation reaction produces 67M-1, 67M-2, and 67M-4, which are pharmacologically active metabolites. 67M-1, 67M-2, and 67M-4 can further undergo glucuronidation and sulfation. Hydroxy metabolites are present in human plasma at much lower concentrations than the parent drug. Biological Half-Life The apparent mean terminal elimination half-life of approximately 5 to 8 hours. |
||
| 毒性/毒理 (Toxicokinetics/TK) |
Hepatotoxicity
Liver test abnormalities have been reported to occur in 2% to 13% (average ~3.5%) of patients receiving febuxostat, but the levels are generally mild-to-moderate and self-limited. The height, nature and timing of these abnormalities have not been described. However, liver test elevations were the major reason for febuxostat discontinuation for adverse events (~2%) in clinical trials, despite the fact that no cases of jaundice or acute hepatitis were reported. Since its approval and more wide-scale use, there have been several individual case reports of liver injury attributed to febuxostat. Most cases have been marked by serum aminotransferase elevations without jaundice arising within days of starting febuxostat, including enzyme elevations in the setting of DRESS syndrome. At least one instance of a mixed-cholestatic hepatitis without immunoallergic features, arising after several months of treatment has been described. The product label for febuxostat lists hepatic steatosis, hepatitis and hepatomegaly as potential side effects. Furthermore, several cases of acute liver failure during febuxostat therapy have been reported to pharmacovigilance databases. Another unrelated, nonpurine xanthine oxidase inhibitor (benzbromarone) was not approved for use in the United States because of its potential for hepatic toxicity. Effects During Pregnancy and Lactation ◉ Summary of Use during Lactation No information is available on the use of febuxostat during breastfeeding. Because febuxostat is more than 99% bound to plasma proteins, the amount in milk is likely to be low. Furthermore, oral bioavailability is only about 50%, so the amount an infant receives systemically is expected to be very small. If febuxostat is required by the mother, it is not a reason to discontinue breastfeeding; however, until more data become available, an alternate drug may be preferred. ◉ Effects in Breastfed Infants Relevant published information was not found as of the revision date. ◉ Effects on Lactation and Breastmilk Relevant published information was not found as of the revision date. Protein Binding Febuxostat is approximately 99.2% bound to plasma proteins, primarily to albumin. Plasma protein binding is constant over the concentration range achieved with 40 mg and 80 mg doses. |
||
| 参考文献 |
|
||
| 其他信息 |
Pharmacodynamics
Febuxostat is a novel, selective xanthine oxidase/dehydrogenase inhibitor that works by decreasing serum uric acid in a dose-dependent manner. In healthy subjects, febuxostat decreased the mean serum uric acid and serum xanthine concentrations, as well as the total urinary uric acid excretion. Febuxostat at daily doses of 40-80 mg reduced the 24-hour mean serum uric acid concentrations by 40 to 55%. Closely related to the drug-induced reduction of serum uric acid levels and mobilization of urate crystals in tissue deposits, febuxostat is associated with gout flares. Unlike [allopurinol] and [oxypurinol], febuxostat has no inhibitory actions against other enzymes involved in purine and pyrimidine synthesis and metabolism, because it does not structurally resemble purines or pyrimidines. Mechanism of Action Gout is a form of acute arthritis that is characterized by the accumulation of crystals of monosodium urate and urate crystals in or around a joint, leading to inflammation and persistent urate crystal deposition in bones, joints, tissues, and other organs that may exacerbate over time. Hyperuricemia is closely related to gout, whereby it may exist for many years before the first clinical attack of gout; thus, aberrated serum uric acid levels and hyperuricemia are believed to be the biochemical aberration involved in the pathogenesis of gout. Xanthine oxidoreductase (XOR) can act as a xanthine oxidase or xanthine dehydrogenase. In humans, it is a critical enzyme for uric acid production as it catalyzes the oxidation reaction steps from hypoxanthine to xanthine and from xanthine to uric acid in the pathway of purine metabolism. Febuxostat potently inhibits XOR, blocking both its oxidase and dehydrogenase activities. With high affinity, febuxostat binds to XOR in a molecular channel leading to the molybdenum-pterin active site, where [allopurinol] demonstrates relatively weak competitive inhibition. XOR is mainly found in the dehydrogenase form under normal physiological conditions; however, in inflammatory conditions, XOR can be converted into the xanthine oxidase form, which catalyzes reactions that produce reactive oxygen species (ROS), such as peroxynitrite. ROS contribute to vascular inflammation and alterations in vascular function. As febuxostat can inhibit both forms of XOR, it can inhibit ROS formation, oxidative stress, and inflammation. In a rat model, febuxostat suppressed renal ischemia-reperfusion injury by attenuating oxidative stress. Febuxostat is a 1,3-thiazolemonocarboxylic acid that is 4-methyl-1,3-thiazole-5-carboxylic acid which is substituted by a 3-cyano-4-(2-methylpropoxy)phenyl group at position 2. It is an orally-active, potent, and selective xanthine oxidase inhibitor used for the treatment of chronic hyperuricaemia in patients with gout. It has a role as an EC 1.17.3.2 (xanthine oxidase) inhibitor. It is an aromatic ether, a nitrile and a 1,3-thiazolemonocarboxylic acid. Febuxostat is a non-purine xanthine oxidase (XO) inhibitor. In early 2008, febuxostat was granted marketing authorization by the European Commission for the treatment of chronic hyperuricemia and gout. In the following year, the FDA for approved febuxostat for use in the chronic management of hyperuricemia in adult patients with gout who have an inadequate response or intolerance to [allopurinol]. Gout is a form of arthritis that is caused by the accumulation of uric acid crystal in or around a joint, leading to inflammation and further deposition of uric acid crystal deposition in bones, joints, tissues, and other organs in the long term. Gout is closely associated with hyperuricemia. Febuxostat works by inhibiting the activity of an enzyme that is responsible for the synthesis of uric acid, thereby reducing serum uric acid levels. In February 2019, a black box warning for febuxostat was added, based on the findings of a post-market clinical study (the CARES trial) where there was an increased risk of cardiovascular (CV) fatal outcomes in patients with gout and known cardiovascular disease treated with febuxostat, when compared to those treated with allopurinol. The manufacturer and the FDA advise health professionals to limit the use of febuxostat to second-line therapy in patients who have inadequate response or intolerance to allopurinol, and to avoid the use of febuxostat in patients with cardiovascular diseases. Febuxostat is a Xanthine Oxidase Inhibitor. The mechanism of action of febuxostat is as a Xanthine Oxidase Inhibitor. Febuxostat is a newly introduced nonpurine xanthine oxidase inhibitor used for the treatment of gout. Chronic febuxostat therapy has been associated with minor serum aminotransferase elevations, but has yet to be linked to cases of clinically apparent acute liver injury. Febuxostat is an orally available, non-purine inhibitor of xanthine oxidase with uric acid lowering activity. Upon oral administration, febuxostat selectively and noncompetitively inhibits the activity of xanthine oxidase, an enzyme that converts oxypurines, including hypoxanthine and xanthine, into uric acid. By inhibiting xanthine oxidase, uric acid production is reduced and serum uric acid levels are lowered. Febuxostat may provide protection against acute renal failure caused by the excessive release of uric acid that occurs upon massive tumor cell lysis resulting from the treatment of some malignancies. FEBUXOSTAT is a small molecule drug with a maximum clinical trial phase of IV (across all indications) that was first approved in 2009 and is indicated for gout and hyperuricemia and has 7 investigational indications. This drug has a black box warning from the FDA. A thiazole derivative and inhibitor of XANTHINE OXIDASE that is used for the treatment of HYPERURICEMIA in patients with chronic GOUT. |
| 分子式 |
C16H16N2O3S
|
|
|---|---|---|
| 分子量 |
316.37
|
|
| 精确质量 |
316.088
|
|
| 元素分析 |
C, 60.74; H, 5.10; N, 8.85; O, 15.17; S, 10.13
|
|
| CAS号 |
144060-53-7
|
|
| 相关CAS号 |
Febuxostat-d9;1246819-50-0;Febuxostat sodium;1140907-13-6;Febuxostat-d7;1285539-74-3
|
|
| PubChem CID |
134018
|
|
| 外观&性状 |
White to off-white solid powder
|
|
| 密度 |
1.3±0.1 g/cm3
|
|
| 沸点 |
536.6±60.0 °C at 760 mmHg
|
|
| 熔点 |
238-239ºC
|
|
| 闪点 |
278.3±32.9 °C
|
|
| 蒸汽压 |
0.0±1.5 mmHg at 25°C
|
|
| 折射率 |
1.606
|
|
| LogP |
4.87
|
|
| tPSA |
111.45
|
|
| InChi Key |
BQSJTQLCZDPROO-UHFFFAOYSA-N
|
|
| InChi Code |
InChI=1S/C16H16N2O3S/c1-9(2)8-21-13-5-4-11(6-12(13)7-17)15-18-10(3)14(22-15)16(19)20/h4-6,9H,8H2,1-3H3,(H,19,20)
|
|
| 化学名 |
2-[3-cyano-4-(2-methylpropoxy)phenyl]-4-methyl-1,3-thiazole-5-carboxylic acid
|
|
| 别名 |
|
|
| HS Tariff Code |
2934.99.9001
|
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
|
|||
|---|---|---|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 2.5 mg/mL (7.90 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 25.0 mg/mL 澄清 DMSO 储备液加入900 μL 玉米油中,混合均匀。 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 3.1609 mL | 15.8043 mL | 31.6086 mL | |
| 5 mM | 0.6322 mL | 3.1609 mL | 6.3217 mL | |
| 10 mM | 0.3161 mL | 1.5804 mL | 3.1609 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
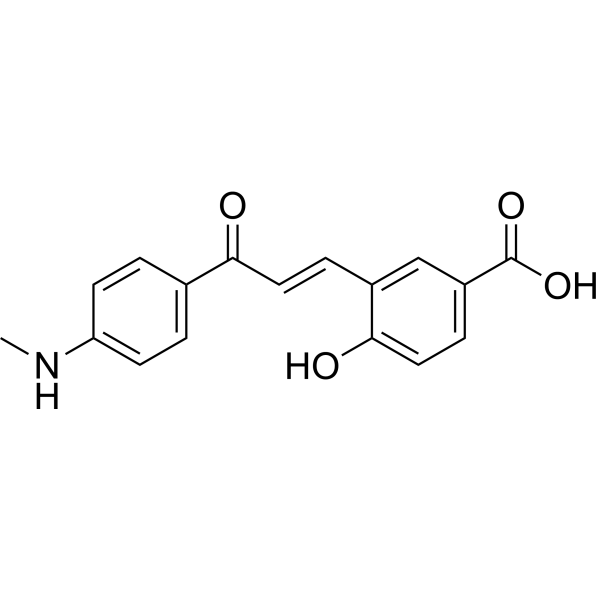 Xanthine oxidase-IN-16
Xanthine oxidase-IN-16
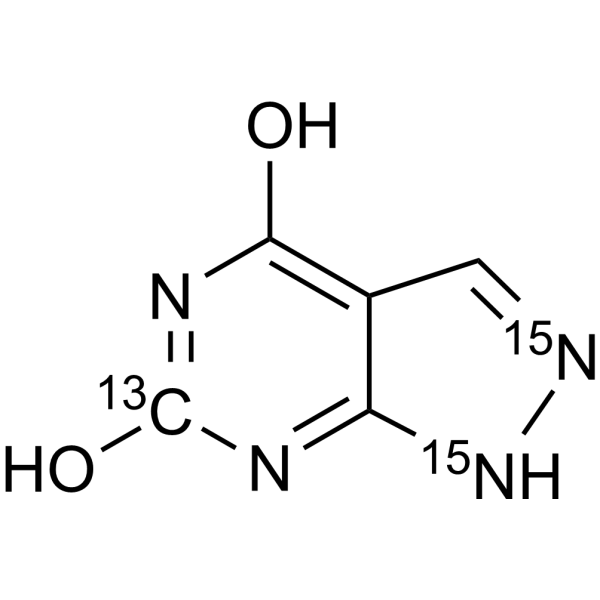 Oxypurinol-13C,15N2
Oxypurinol-13C,15N2
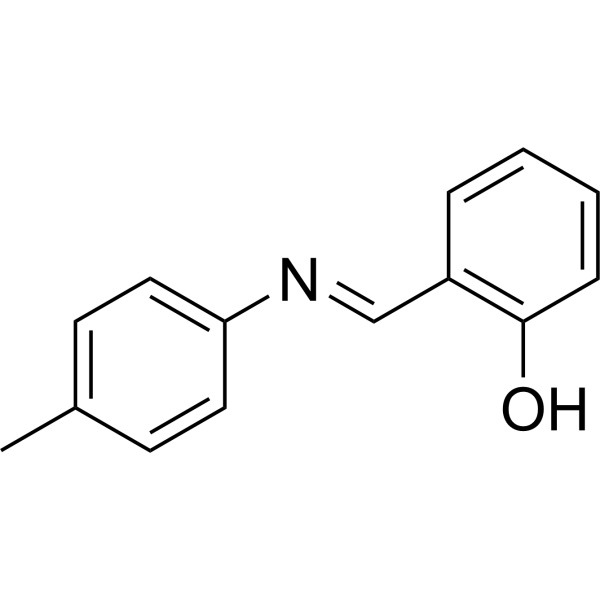 Xanthine oxidase-IN-13
Xanthine oxidase-IN-13
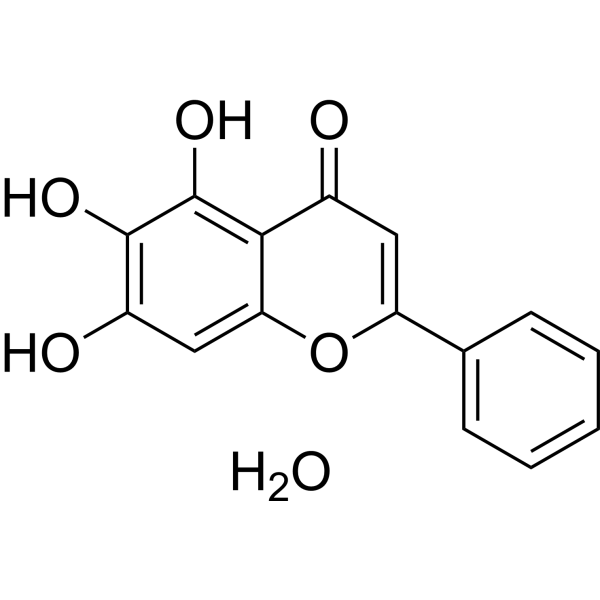 Baicalein hydrate
Baicalein hydrate
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1





























 COA
COA
