| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 5mg |
|
||
| 10mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| Other Sizes |
|
| 靶点 |
Carnitine palmitoyltransferase I (CPT-I)
|
|---|---|
| 体外研究 (In Vitro) |
当依托莫克不可逆地结合到 CPT-1 的催化位点时,它会抑制 CPT-1 的活性,同时增加移植氧化酶。 Etomoxir 被创建为线粒体外膜定位的线粒体肉毒碱支架扩增酶-1 (CPT-1) 的探针。 Etomoxir 通过充当氧化体增殖剂来刺激心肌中的 DNA 合成和心肌发育。因此,etomoxan 被视为除 CPT1 之外的 PPARα 激动剂 [1]。依托莫克西已被提议作为激活心脏突变的靶标。它是环氧乙烷激酶肉毒碱模板转移酶 I 家族的成员。肉毒碱模板转移酶 I 活性在依托莫克西治疗激活后不可逆地转录。因此,线粒体和β-氧化的碱基输入减少,导致细胞质积累和氧化增加。 Etomoxir 的长时间延迟(24 小时)对酶的表达甚至有明显的影响 [2]。
|
| 体内研究 (In Vivo) |
Etomoxir 是一种参与自由平衡 (FFA) 氧化的转录因子,并与重要的酶 CPT1 相关。 P53 直接响应 Bax,而 Bax 被依托莫克西阻断,这一事实进一步支持了 P53 和 Bax 的直接响应,以及在 db/db 模型中FAO 介导的催化 ROS 产生的作用。以 20 mg/kg 体重的剂量,每天注射一次依托莫克西,持续八天,导致特异性 CPT-I 活性降低 44%。在用依托莫克西处理的催化剂中,催化剂CPT-I活性降低了44%。使用 20 mg/kg Etomoxir 治疗 8 天后,Lewis 的血糖水平保持不变,与之前的 Etomoxir 喂养试验一致。同样,依托莫克西的喂养对后肢的收获质量或体重增加等一般生长性状没有影响。然而,在接受依托莫克西治疗的患者中,肝脏和心脏质量均显着增加了 11% [4]。
Etomoxir 可抑制 BMSC 分化的成骨细胞的减少,并显着抑制高脂肪 (HF) 和 db/db 饮食喂养的小鼠骨矿物质密度 (BMD) 和破骨强度的下降[3]。在喂食 HF 和 db/db 的小鼠中,依托泊昔抑制成骨细胞和小鼠线粒体 ROS 生成的增加[3]。依托莫克舍引起的体内肉碱棕榈酰转移酶-I (CPT-I) 部分抑制不会改变心脏长链脂肪酸的摄取和氧化速率[4]。 本研究评估了2型糖尿病患者游离脂肪酸(FFA)、ROS产生、线粒体功能障碍与骨密度(BMD)之间的关联,并探讨了其分子机制。db/db和高脂肪(HF)喂养的小鼠接受CPT1、MitoQ抑制剂Etomoxir和P53抑制剂PFT-α的治疗。评估骨代谢因素,分离BMSCs并诱导成骨分化。2型糖尿病患者的FFA、脂质过氧化和mtDNA拷贝数与BMD相关Etomoxir、MitoQ和PFT-α显著抑制了db/db和HF喂养的小鼠BMD和骨断裂强度的降低,并抑制了BMSCs分化成骨细胞的减少。Etomoxir和MitoQ,而不是PFT-α,抑制了db/db和HF喂养的小鼠和成骨细胞线粒体ROS产生的增加。此外,Etomoxir、MitoQ和PFT-α显著抑制了成骨细胞的线粒体功能障碍。此外,db/db和HF喂养的小鼠成骨细胞中的线粒体凋亡被激活,而Etomoxir、MitoQ和PFT-α则抑制了线粒体凋亡。此外,P53的线粒体积累募集Bax并引发凋亡事件的分子事件。这些结果表明,脂肪酸氧化导致ROS产生,激活P53/Bax介导的线粒体凋亡,导致T2DM成骨分化和骨丢失减少。[3] 尽管CPT-I(肉碱棕榈酰基转移酶-I)通常被认为是线粒体β氧化的主要速率控制位点,但CPT-I是否在心脏的整体LCFA(长链脂肪酸)通量中起限速作用尚不完全清楚。心脏中调节LCFA通量的另一个重要部位是CD36和FABPpm(质膜脂肪酸结合蛋白)促进的跨肌膜LCFA转运。因此,我们探讨了在LCFA-CoA线粒体进入水平上对LCFA通量的慢性药理学阻断在多大程度上会影响肌膜LCFA摄取。每天给大鼠注射生理盐水或依托莫西(一种特定的CPT-I抑制剂),持续8天,剂量为20mg/kg体重。etomoxir治疗的大鼠心脏CPT-I活性降低了44%。与对照组相比,依托莫西治疗的大鼠心脏中CD36和FABPpm的肌浆含量以及LCFA转运能力没有改变。此外,无论是在基础代谢需求下还是在急性诱导的最大代谢需求下,依托莫西治疗大鼠的LCFA摄取和氧化率以及心肌细胞对葡萄糖的摄取率与对照组大鼠没有差异。最后,依托莫西治疗大鼠的心脏没有显示出三酰甘油的积累。因此,CPT-I似乎不是心脏总LCFA通量的主要心率控制位点。肌膜LCFA进入而非线粒体LCFA-CoA进入可能是心脏代谢疾病中LCFA通量正常化的有前景的靶点[4]。 |
| 酶活实验 |
生化测量[3]
血样在4°C下以3000rpm离心10分钟。根据制造商的说明,使用ELISA试剂盒测量血清FFA。用商品试剂盒检测血清甘油三酯、高密度脂蛋白和低密度脂蛋白。使用Elisa测定试剂盒测量血清碱性磷酸酶(ALP)、骨钙素(OCN)和酒石酸抗性酸性磷酸酶(TRAP)以评估成骨和破骨细胞活性。 脂质过氧化的测量[3] 使用Histopaque-1119和Histopaque-1077通过等密度离心分离和纯化PBMC。根据制造商的说明,使用商业试剂盒通过测量硫代巴比妥酸反应物质(TBARS)水平来评估PBMC和分化成骨细胞中的脂质过氧化。 |
| 细胞实验 |
我们检测了依托莫治疗对H9c2心肌成肌细胞中从头心磷脂(CL)生物合成的影响。Etomoxir处理不影响CL生物合成和重塑酶的活性,但导致[1-14C]棕榈酸或[1-14C]油酸掺入CL的减少。其机制是通过利用膜磷脂酸磷酸水解酶活性增加35%(P<0.05)介导的反应,将脂质合成重定向为1,2-二酰基-sn-甘油,通过CL生物合成的从头途径减少脂肪酸流量。相反,依托莫司处理增加了[1,3H]甘油掺入CL。其机制是甘油激酶活性增加33%(P<0.05),这通过CL生物合成的从头途径产生了增加的甘油流量。Etomoxir处理抑制了81%的1,2-二酰基-sn-甘油酰基转移酶活性(P<0.05),从而引导甘油和脂肪酸从1,2,3-三酰基-sn-胆固醇的利用转向磷脂酰胆碱和磷脂酰乙醇胺的生物合成。相反,依托莫西抑制了肌醇与磷脂酰肌醇的结合,其机制是抑制肌醇的摄取。Etomoxir不影响[3H]丝氨酸的摄取,但导致衍生自磷脂酰丝氨酸的磷脂酰乙醇胺的形成增加。结果表明,依托莫西处理对不同代谢前体的甘油从头生物合成具有不同的影响。此外,依托莫西介导甘油和脂肪酸前体进入CL的独特和差异的代谢通道[2]。
|
| 动物实验 |
Animal treatment[3]
All animal experiments were performed according to the procedures approved by Fourth Military Medical University Animal Care and Use Committee and were carried out in accordance with the approved guidelines. 80 male C57BLKS/J lar-Leprdb/db mice and 20 wild type littermates (8 week) were obtained from Model Animal Research Centre, Nanjing University, China. Mice were housed in cages in a limited access room, under temperature (23 ± 2 °C) and humidity (55 ± 5%) condition with a standard light (12 h light/dark) cycle and fed a regular diet. db/db mice were randomly divided into four groups: db/db group, Etomoxir group, MitoQ group, and PFT-α group. In the Etomoxir group, mice were intraperitoneally injected with 1 mg/kg Etomoxir twice every week. In the MitoQ group, 50 μmol/L MitoQ was given to the mice in water. Water bottles, containing either MitoQ, were covered with aluminum foil, and all bottles were refilled every 3 days. In the PFT-α group, mice were intraperitoneally injected with 1 mg/kg PFT-α twice every week. WT mice were administrated with vehicle instead. The experimental period is 8 weeks. At the end, peripheral blood samples and bone marrow cells were harvested for the assays. 100 C57BL/6 mice obtained from Experimental Animal Centre of Fourth Military Medical University. The mice were randomly divided into five groups: Control group, HF diet group, Etomoxir group, MitoQ group, and PFT-α group. Mice in HF diet, Etomoxir, MitoQ, and PFT-α groups were given high fat diet for 20 weeks and mice in Etomoxir, MitoQ, and PFT-α groups were administrated with Etomoxir, MitoQ, and PFT-α in the last 10 weeks. The administration of Etomoxir, MitoQ, and PFT-α were identical to the treatment in db/db mice. Control mice were administrated with vehicle instead. Rats were injected daily with saline or etomoxir, a specific CPT-I inhibitor, for 8 days at 20 mg/kg of body mass. Etomoxir-treated rats displayed a 44% reduced cardiac CPT-I activity. Sarcolemmal contents of CD36 and FABPpm, as well as the LCFA transport capacity, were not altered in the hearts of etomoxir-treated versus control rats. Furthermore, rates of LCFA uptake and oxidation, and glucose uptake by cardiac myocytes from etomoxir-treated rats were not different from control rats, neither under basal nor under acutely induced maximal metabolic demands. Finally, hearts from etomoxir-treated rats did not display triacylglycerol accumulation. Therefore CPT-I appears not to present a major rate-controlling site in total cardiac LCFA flux. It is likely that sarcolemmal LCFA entry rather than mitochondrial LCFA-CoA entry is a promising target for normalizing LCFA flux in cardiac metabolic diseases.[4] |
| 参考文献 |
|
| 其他信息 |
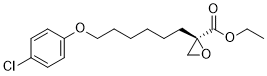
(2R)-2-[6-(4-chlorophenoxy)hexyl]-2-oxiranecarboxylic acid ethyl ester is an aromatic ether.
Partial fatty acid oxidation inhibitors have raised great interest since they are expected to counteract a dysregulated gene expression of hypertrophied cardiocytes. Some of these compounds have been developed for treating non-insulin-dependent diabetes mellitus and stable angina pectoris. A shift from fatty acid oxidation to glucose oxidation leads to a reduced gluconeogenesis and improved economy of cardiac work. An increased glucose oxidation can be achieved with the following enzyme inhibitors: etomoxir, oxfenicine, methyl palmoxirate, S-15176, metoprolol, amiodarone, perhexiline (carnitine palmitoyltransferase-1); aminocarnitine, perhexiline (carnitine palmitoyltransferase-2); hydrazonopropionic acid (carnitine-acylcarnitine translocase); MET-88 (gamma-butyrobetaine hydroxylase); 4-bromocrotonic acid, trimetazidine, possibly ranolazine (thiolases); hypoglycin (butyryl-CoA dehydrogenase); dichloroacetate (pyruvate dehydrogenase kinase). CLINICAL TRIALS with trimetazidine and ranolazine showed that this shift in substrate oxidation has an antianginal action. Etomoxir and MET-88 improved the function of overloaded hearts by increasing the density of the Ca(2+) pump of sarcoplasmic reticulum (SERCA2). The promoters of SERCA2 and alpha-myosin heavy-chain exhibit sequences which are expected to respond to transcription factors responsive to glucose metabolites and/or peroxisome proliferator-responsive element (PPAR) agonists. Further progress in elucidating novel compounds which upregulate SERCA2 expression is closely linked to the characterization of regulatory sequences of the SERCA2 promoter. [1] We examined the effect of etomoxir treatment on de novo cardiolipin (CL) biosynthesis in H9c2 cardiac myoblast cells. Etomoxir treatment did not affect the activities of the CL biosynthetic and remodeling enzymes but caused a reduction in [1-14C]palmitic acid or [1-14C]oleic acid incorporation into CL. The mechanism was a decrease in fatty acid flux through the de novo pathway of CL biosynthesis via a redirection of lipid synthesis toward 1,2-diacyl-sn-glycerol utilizing reactions mediated by a 35% increase (P < 0.05) in membrane phosphatidate phosphohydrolase activity. In contrast, etomoxir treatment increased [1,3-3H]glycerol incorporation into CL. The mechanism was a 33% increase (P < 0.05) in glycerol kinase activity, which produced an increased glycerol flux through the de novo pathway of CL biosynthesis. Etomoxir treatment inhibited 1,2-diacyl-sn-glycerol acyltransferase activity by 81% (P < 0.05), thereby channeling both glycerol and fatty acid away from 1,2,3-triacyl-sn-glycerol utilization toward phosphatidylcholine and phosphatidylethanolamine biosynthesis. In contrast, etomoxir inhibited myo-[3H]inositol incorporation into phosphatidylinositol and the mechanism was an inhibition in inositol uptake. Etomoxir did not affect [3H]serine uptake but resulted in an increased formation of phosphatidylethanolamine derived from phosphatidylserine. The results indicate that etomoxir treatment has diverse effects on de novo glycerolipid biosynthesis from various metabolic precursors. In addition, etomoxir mediates a distinct and differential metabolic channeling of glycerol and fatty acid precursors into CL. [2] This study evaluated the association between free fatty acid (FFA), ROS generation, mitochondrial dysfunction and bone mineral density (BMD) in type 2 diabetic patients and investigated the molecular mechanism. db/db and high fat (HF)-fed mice were treated by Etomoxir, an inhibitor of CPT1, MitoQ, and PFT-α, an inhibitor of P53. Bone metabolic factors were assessed and BMSCs were isolated and induced to osteogenic differentiation. FFA, lipid peroxidation and mtDNA copy number were correlated with BMD in T2DM patients. Etomoxir, MitoQ and PFT-α significantly inhibited the decrease of BMD and bone breaking strength in db/db and HF-fed mice and suppressed the reduction of BMSCs-differentiated osteoblasts. Etomoxir and MitoQ, but not PFT-α, inhibited the increase of mitochondrial ROS generation in db/db and HF-fed mice and osteoblasts. In addition, Etomoxir, MitoQ and PFT-α significantly inhibited mitochondrial dysfunction in osteoblasts. Moreover, mitochondrial apoptosis was activated in osteoblasts derived from db/db and HF-fed mice, which was inhibited by Etomoxir, MitoQ and PFT-α. Furthermore, mitochondrial accumulation of P53 recruited Bax and initiated molecular events of apoptotic events. These results demonstrated that fatty acid oxidation resulted in ROS generation, activating P53/Bax-mediated mitochondrial apoptosis, leading to reduction of osteogenic differentiation and bone loss in T2DM. [3] Although CPT-I (carnitine palmitoyltransferase-I) is generally regarded to present a major rate-controlling site in mitochondrial beta-oxidation, it is incompletely understood whether CPT-I is rate-limiting in the overall LCFA (long-chain fatty acid) flux in the heart. Another important site of regulation of the LCFA flux in the heart is trans-sarcolemmal LCFA transport facilitated by CD36 and FABPpm (plasma membrane fatty acid-binding protein). Therefore, we explored to what extent a chronic pharmacological blockade of the LCFA flux at the level of mitochondrial entry of LCFA-CoA would affect sarcolemmal LCFA uptake. Rats were injected daily with saline or etomoxir, a specific CPT-I inhibitor, for 8 days at 20 mg/kg of body mass. Etomoxir-treated rats displayed a 44% reduced cardiac CPT-I activity. Sarcolemmal contents of CD36 and FABPpm, as well as the LCFA transport capacity, were not altered in the hearts of etomoxir-treated versus control rats. Furthermore, rates of LCFA uptake and oxidation, and glucose uptake by cardiac myocytes from etomoxir-treated rats were not different from control rats, neither under basal nor under acutely induced maximal metabolic demands. Finally, hearts from etomoxir-treated rats did not display triacylglycerol accumulation. Therefore CPT-I appears not to present a major rate-controlling site in total cardiac LCFA flux. It is likely that sarcolemmal LCFA entry rather than mitochondrial LCFA-CoA entry is a promising target for normalizing LCFA flux in cardiac metabolic diseases. [4] |
| 分子式 |
C17H23CLO4
|
|---|---|
| 分子量 |
326.82
|
| 精确质量 |
326.128
|
| 元素分析 |
C, 62.48; H, 7.09; Cl, 10.85; O, 19.58
|
| CAS号 |
124083-20-1
|
| 相关CAS号 |
Etomoxir sodium salt;828934-41-4; 82258-36-4 (racemate) 124083-20-1 (free acid); 828934-40-3 (S-isomer); 132308-39-5 (potassium salt)
|
| PubChem CID |
9840324
|
| 外观&性状 |
Colorless to light yellow solid (<32°C),or liquid (>34°C)
|
| 密度 |
1.2±0.1 g/cm3
|
| 沸点 |
405.0±25.0 °C at 760 mmHg
|
| 闪点 |
142.6±22.2 °C
|
| 蒸汽压 |
0.0±0.9 mmHg at 25°C
|
| 折射率 |
1.520
|
| LogP |
4.46
|
| tPSA |
48.06
|
| 氢键供体(HBD)数目 |
0
|
| 氢键受体(HBA)数目 |
4
|
| 可旋转键数目(RBC) |
11
|
| 重原子数目 |
22
|
| 分子复杂度/Complexity |
342
|
| 定义原子立体中心数目 |
1
|
| SMILES |
O=C(OCC)[C@@]1(OC1)CCCCCCOC2=CC=C(C=C2)Cl
|
| InChi Key |
DZLOHEOHWICNIL-QGZVFWFLSA-N
|
| InChi Code |
InChI=1S/C17H23ClO4/c1-2-20-16(19)17(13-22-17)11-5-3-4-6-12-21-15-9-7-14(18)8-10-15/h7-10H,2-6,11-13H2,1H3/t17-/m1/s1
|
| 化学名 |
Ethyl (2R)-2-[6-(4-chlorophenoxy)hexyl]oxirane-2-carboxylate
|
| 别名 |
(R)-(+)-Etomoxir B 807-54 B80754 B-80754B807-54 B-807-54 B 80754
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month 注意: (1). 本产品在运输和储存过程中需避光。 (2). 请将本产品存放在密封且受保护的环境中(例如氮气保护),避免吸湿/受潮。 |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
DMSO : ~100 mg/mL (~305.98 mM)
|
|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: 3.5 mg/mL (10.71 mM) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 悬浮液;超声助溶。
例如,若需制备1 mL的工作液,可将100 μL 35.0mg/mL澄清的DMSO储备液加入到900μL 20%SBE-β-CD生理盐水中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 配方 2 中的溶解度: ≥ 3.5 mg/mL (10.71 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 35.0 mg/mL 澄清 DMSO 储备液加入到 900 μL 玉米油中并混合均匀。 View More
配方 3 中的溶解度: 2.5 mg/mL (7.65 mM) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液; 超声助溶. 配方 4 中的溶解度: 10 mg/mL (30.60 mM) in 0.5% Methylcellulose/saline water (这些助溶剂从左到右依次添加,逐一添加), 悬浊液; 超声助溶。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 3.0598 mL | 15.2989 mL | 30.5979 mL | |
| 5 mM | 0.6120 mL | 3.0598 mL | 6.1196 mL | |
| 10 mM | 0.3060 mL | 1.5299 mL | 3.0598 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
| NCT Number | Recruitment | interventions | Conditions | Sponsor/Collaborators | Start Date | Phases |
| NCT03144128 | COMPLETED | Dietary Supplement: Vitamin D Dietary Supplement: Placebo |
Cancer Cachexia Vitamin D Deficiency |
David Travis Thomas | 2018-05-23 | Not Applicable |
|
|
|
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1






























 COA
COA







